 夜雨聆风
夜雨聆风





设计和开发要求形成的文件(医疗器械)
课程推荐↓
1、前言
《医疗器械生产质量管理规范 附录体外诊断试剂现场检查指导原则》国药总局(2015年第103号)第5章对医疗器械设计和开发工作做出了明确的规定。
按照《指导原则》,设计和开发工作可分为:策划、输入、输出、转换和验证、确认,以及设计和开发更改等阶段。
现在,我们给大家梳理一下设计和开发工作要求形成的文件:
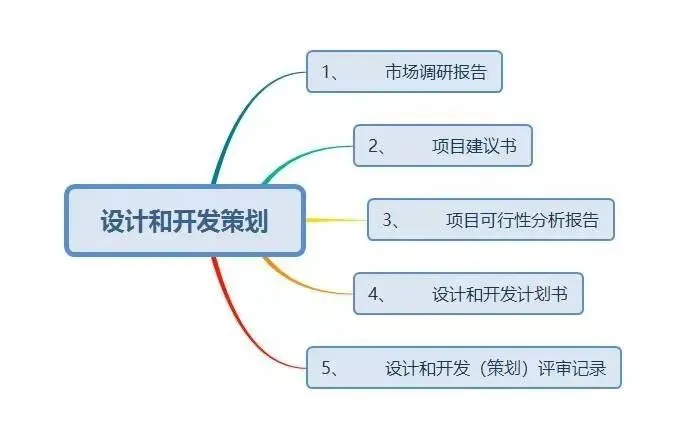
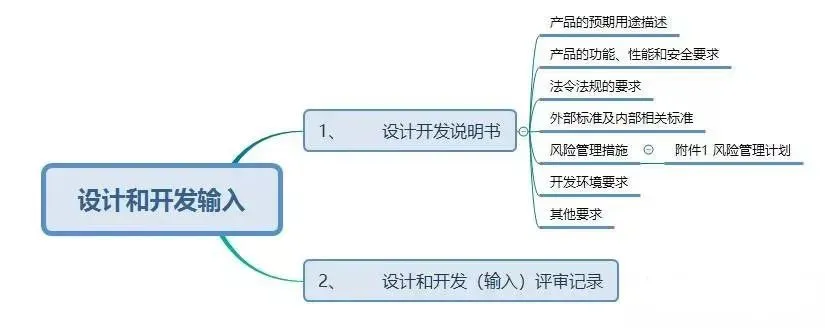
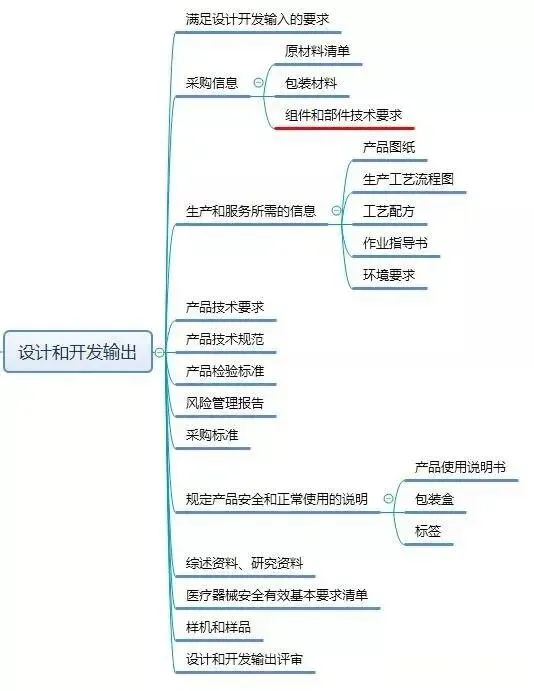
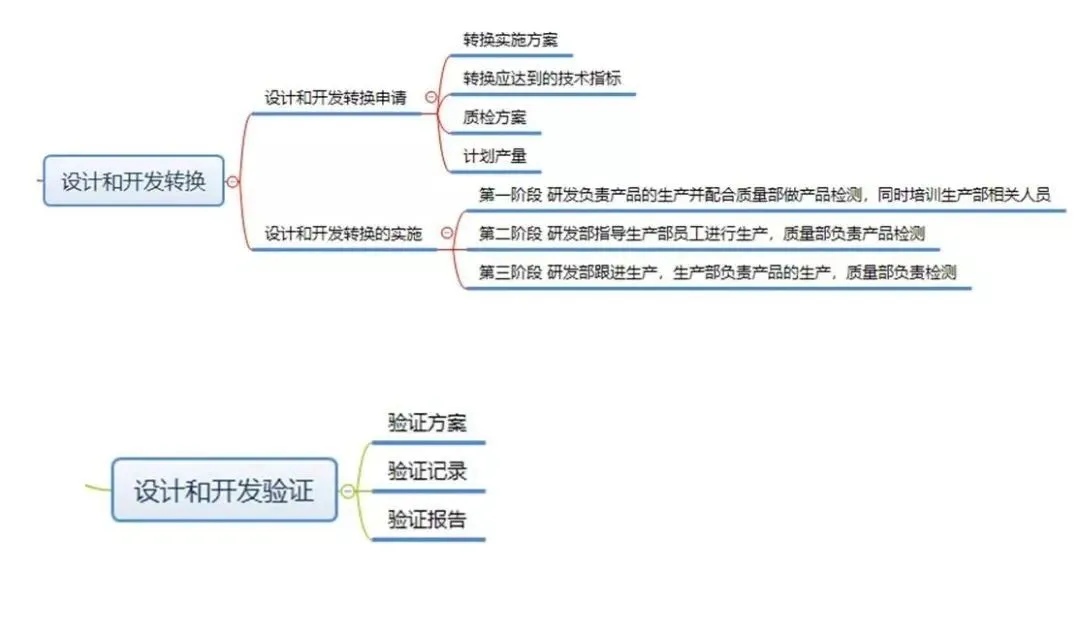
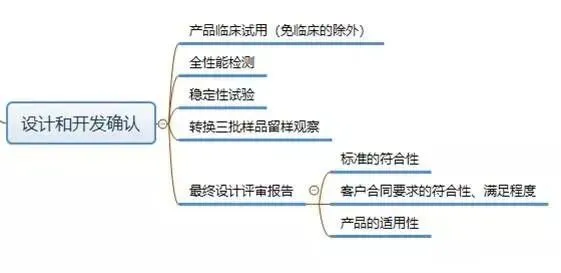
结束语:
设计和开发工作除了以上要求形成的文件以外,还应保留设计和开发期间相应的过程记录。
此外,若在设计和开发期间发生更改,还应保留设计和开发更改的相关记录。并对设计和开发更改进行相应的评审和风险风险分析。
— 往期回顾 —
近期培训课题安排
请点击链接查看详细内容
●11月18-22日上海实操2020版《中国药典》无菌微生物检(化)验员培训班
●11月25-29日杭州实操2020版《中国药典》无菌微生物检(化)验员培训班
●11月28-29日线上医疗器械质量管理体系内审员培训班(网课)
●12月12-13日线上医疗器械生产质量管理规范解读及案例解析(网课)
●12月17-19杭州医疗器械品设计开发与注册申报
●12月2-6日北京实操2020版《中国药典》无菌微生物检(化)验员培训班
●12月9-13日苏州实操2020版《中国药典》无菌微生物检(化)验员培训班
●12月24-26杭州医疗器械可用性涉及、测试、合规实战
如需正式红头文件及报名咨询:
培训中心负责人:位宁18611200350(微信同号)
咨询QQ:787116116
实操课程人员有限,如需请尽快联系报名
如需其他课题学习或内训均可联系~烦请分享转发


识别左侧二维码
加位宁咨询
