 夜雨聆风
夜雨聆风





跟着Nature源代码学做图丨系统发育树 * 功能热图联合可视化


Nature | 文献

https://www.nature.com/articles/s41586-023-06990-w文章 Figure 1d
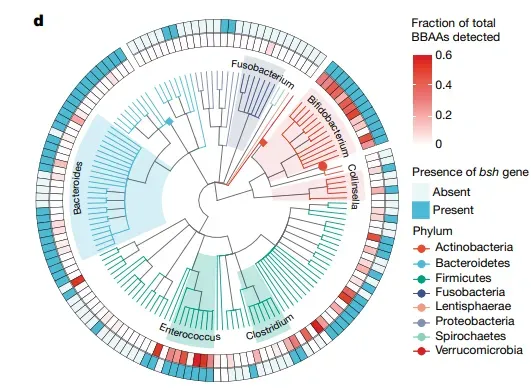
以系统发育角度展示各菌株的“BBAA 产能”差异。
外环颜色(白 → 红)表示该菌株能产生多少种 BBAA。红色越深,说明它能合成更多不同类型的 BBAA。
用内环标记每个菌株是否含有 bsh 基因。
蓝色 = 缺失 bsh
红色 = 含有 bsh
Bifidobacterium、Enterococcus:大量携带 bsh,BBAA 产量高。
Fusobacterium:缺乏 bsh,几乎不产生 BBAA。
Bacteroides:部分携带 bsh,产能中等。
文章源代码

GitHub - PattersonLab-PSU/BBAAs-paper
在 figure1d 中,跟着文章的源代码画图
Rstudio | 画图
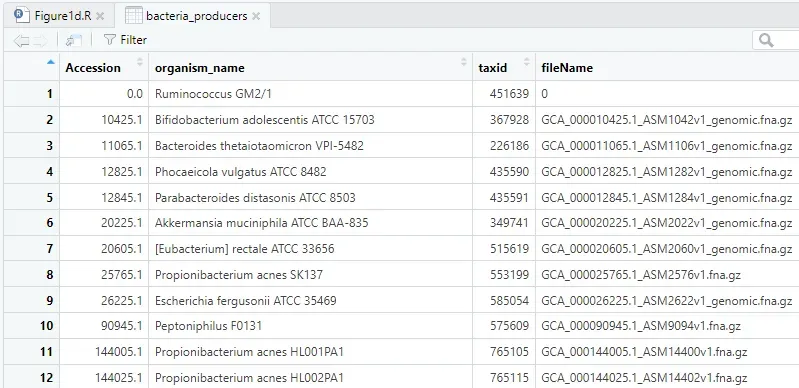
bacteria_producers<- read.csv("producer_gene.csv") %>%select(-X) %>%distinct(Accession, .keep_all = TRUE) %>%rename(fileName=label) %>%mutate(Accession=as.numeric(gsub("GC[F|A]_","",Accession))) %>%select(-gene,-n)
-
去掉多余列
X,并按Accession去重。 -
label改名为fileName(可能是后面要和文件名对应)。 -
把
Accession从类似GCF_000123这种文本,去掉前缀GCF_/GCA_再转成数值。 -
去掉
gene和n两列(后面不画,只留下每个菌的 MCBA 相关汇总信息)。
prokka_gene_presence_absence <- read.csv("prokka_merged_output.csv",header=TRUE) %>%rename(Accession="locus_tag") %>%mutate(Accession=gsub("GC[A|F]_","",Accession),Accession=as.numeric(Accession)) %>%select(-X)
-
locus_tag改名成Accession。 -
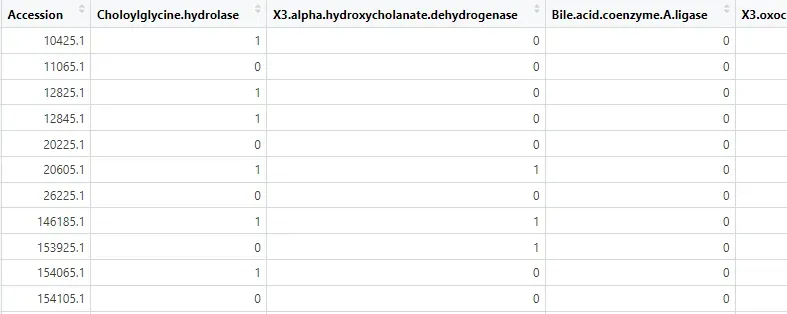
这张表里有一列
Choloylglycine.hydrolase,代表 bsh 基因 presence/absence(0/1)。
bacteria_taxonomy<- read.csv("merged_metadata.csv")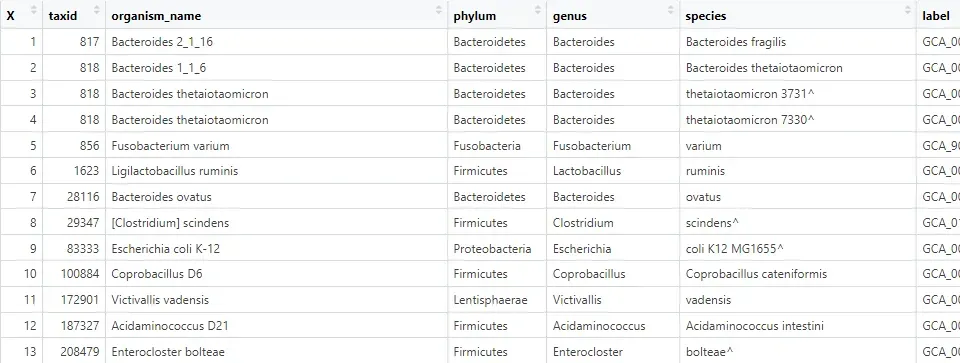
tree<-ggtree::read.tree("phyliptree.ph")-
phyliptree.ph:研究中这些菌株构建好的系统发育树。
tree_taxid <- read.table("read_tree_taxid.txt",sep="\t", header=TRUE) %>%select(name,taxid)
-
read_tree_taxid.txt:树的 tip 名称与taxid的对应表(因为树文件里可能是怪名字,需要转成可与 metadata 对上号的 taxid)。
bacteria_producers<-bacteria_producers %>%merge(prokka_gene_presence_absence,by="Accession",all=TRUE)
把 MCBA 生产信息(producer_gene)和 基因存在矩阵(prokka)按 Accession 合并,后面就可以在同一张图里同时用到:
-
proportion,nMCB(实验测得 MCBA 的比例、数量) -
Choloylglycine.hydrolase(bsh 基因是否存在)
#用 midpoint rooting 给树选一个中点做 root,保证树更“平衡”。tree_rooted <- phangorn::midpoint(tree, node.labels="support")
tree_df <-tree %>% as.tibble %>% mutate(label=gsub("'","",label))-
把树转成 tibble,方便后面合并。
-
把 label 里的
'去掉(有些 newick 文件 tip 名会带')。
tree_df_taxid <- merge(tree_df,tree_taxid, by.x="label", by.y="name") %>%merge(bacteria_taxonomy,by="taxid") %>%select(-c(X,label.y)) %>%rename(label=label.x) %>%mutate(organism_name=gsub("","",organism_name)) %>%mutate(Accession=as.numeric(gsub("GC[F|A]_","",Accession)))
这一步把系统发育树上的每个 tip 和真实的菌信息挂接起来:
-
tree_df+tree_taxid:用树上的label对应到taxid。 -
然后再 +
bacteria_taxonomy:用taxid查到 phylum、genus、organism_name、Accession 等。 -
清理一下列名和前缀,最终得到
tree_df_taxid:
-
每个 tip 都有:
label(树上的名字)、taxid、phylum、genus、Accession等。这就是后面%<+%绑定给树用的 meta data。
tree_rooted$node.label = gsub("'","",tree_rooted$node.label)p <- ggtree(tree_rooted, layout="circular", size=0.5, open.angle=5,branch.length = "none", aes(color=phylum)) %<+%tree_df_taxid +scale_color_npg(palette=c("nrc"),alpha=1,na.value="grey40")
-
layout="circular":画一个环形系统发育树。 -
branch.length="none":不按真实 branch length,所有分支长度统一。 -
%<+% tree_df_taxid:把tree_df_taxid作为 tip 的 meta 信息绑定进去,这样aes(color=phylum)就能按门来上色。 -
scale_color_npg("nrc"):用ggsci的 Nature Publishing Group 调色板。
这一步得到一个彩色环形树,每条叶子(菌株)线段颜色代表它的门。
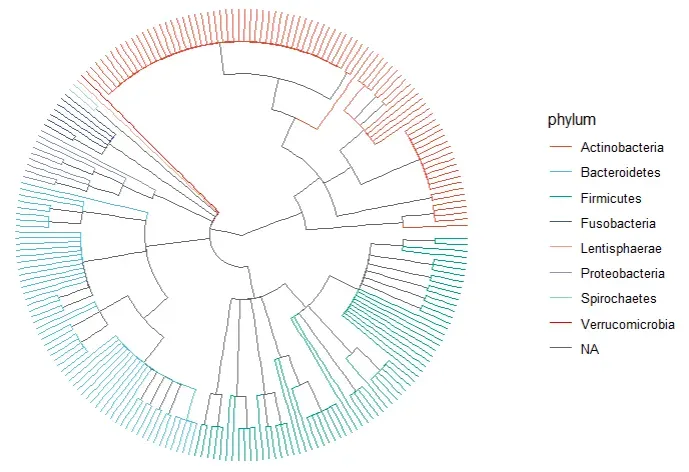
p1 <- p %>% collapse(node=240) + geom_point2(aes(subset=(node==240)),shape=16,size=3)p1 <- p1 %>% collapse(node=236) + geom_point2(aes(subset=(node==236)),shape=18,size=3)p1 <- p1 %>% collapse(node=278) + geom_point2(aes(subset=(node==278)),shape=18,size=3)
-
collapse(node=...):把某个内部节点下的一个大支系折叠成一个三角形、扇形,减少视觉复杂度。 -
geom_point2(subset=(node==...)):在折叠点 mark 一个点(不同形状),表示“这里有折叠”。
目的是:树太复杂时,把不关注的 clade 折叠起来,突出整体结构而不是每个 tip。
tree_df_taxid(树上每个菌的信息)和 bacteria_producerstree_df_producer_gene<-tree_df_taxid %>%merge(bacteria_producers, by=c("Accession","organism_name"), all.x=TRUE) %>%mutate(production = ifelse(nMCB>0,1,0))
-
这样每个 tip 都知道自己是否在实验中被检测过,检测到多少种 MCBAs (
nMCB),以及proportion。 -
production = ifelse(nMCB>0,1,0):有检测到 MCBAs 的标记为 1,否则 0。
proportions_df <- tree_df_producer_gene %>%select(label,proportion) %>%column_to_rownames("label")
label 为行名,仅含 proportion 的矩阵:这正是 gheatmap() 需要的格式:行 = 树的 tip,列 = 要加的 heatmap 值。p2<-gheatmap(p1,proportions_df,offset=1, width=0.1,colnames=FALSE) +scale_fill_gradient2(low = "white",mid="#ED7F7F",high = "#DC0000FF",midpoint = 0.3) +labs(fill="Proportion of MCBAs <br /> detected among tested") +theme(legend.title = element_markdown())
-
gheatmap():在树外围加一圈 heatmap(相当于一个环形条带)。 -
offset=1:与树的距离。 -
width=0.1:这一圈的厚度。 -
scale_fill_gradient2(): -
low = 白色
-
mid / high = 两个红色深浅值
-
midpoint = 0.3:0.3 作为中间值。 -
legend title 用
element_markdown()支持<br />换行。
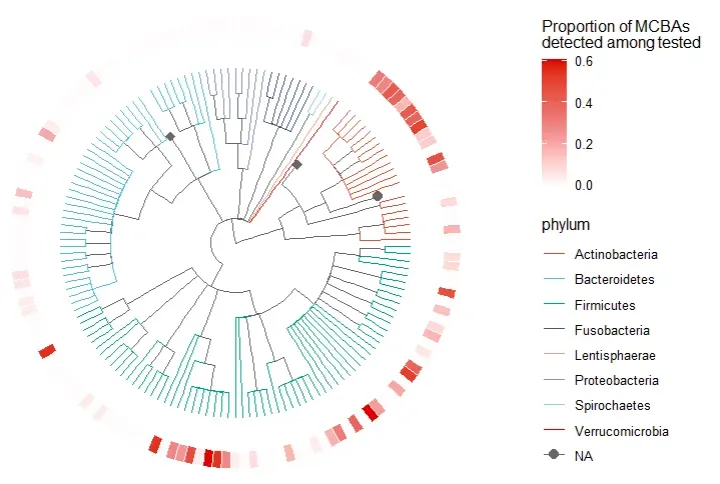
这一圈直观表达:每个菌株在实验中检测到 MCBAs 的比例有多高,颜色越深,MCBAs 产生能力越强。
6.1将0/1转为因子 bsh_df <- tree_df_producer_gene %>%select(label,`Choloylglycine.hydrolase`) %>%column_to_rownames("label") %>%rename(bsh="Choloylglycine.hydrolase") %>%mutate(bsh=case_when(bsh==1 ~ "Present",bsh ==0 ~ "Absent")) %>%mutate(bsh = as.factor(bsh))
-
从合并表里取出
Choloylglycine.hydrolase这一列,表示 bsh 基因是否存在(0/1)。 -
重命名为
bsh,并把 0/1 转为"Present" / "Absent"因子。
gheatmapp3 <- p2 + ggnewscale::new_scale_fill()p4 <- gheatmap(p3,bsh_df,offset=1.75, width=0.1,colnames=FALSE) +scale_fill_manual(values=c("#EBF7FA","#4DBBD5FF"),name="*bsh* gene") +theme(legend.title = element_markdown())
-
ggnewscale::new_scale_fill():因为前面已经用过一次fill(MCBA 比例),要再加一圈新的 fill,需要重启一个新的 fill 颜色空间。 -
第二次
gheatmap(): -
offset=1.75:比第一圈更外面一点。 -
scale_fill_manual():两种颜色分别代表Absent/Present,legend名称用markdown把bsh斜体。
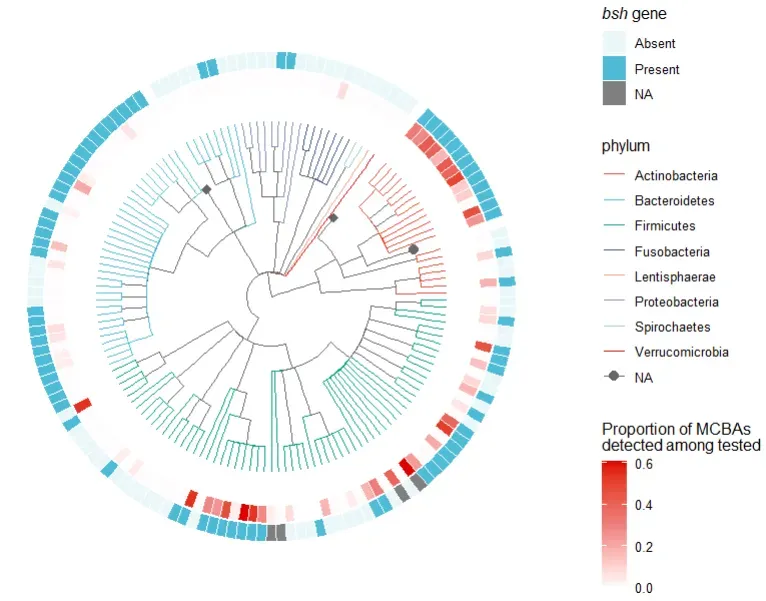
这一圈表达:每个菌株是否携带bsh基因,与内圈的MCBA产生比例可以直观对照,判断 bsh presence 和功能表型的一致性/不一致性。
中心的圆形树展示了:
-
每个枝条代表一个细菌物种
-
树的分叉表示它们之间的 进化距离与祖先关系
分支越接近说明:
-
它们进化上更接近
-
具有潜在相似的基因组特征或生态位偏好
Firmicutes(绿色树枝)中有大量深蓝色 → 说明该门普遍携带 bsh
Bacteroidetes(浅蓝色树枝)中 bsh 也分布较多
Verrucomicrobia(红色)部分缺失 bsh
nodeids <- nodeid(tree, tree_rooted$node.label[tree_rooted$node.label %in% tree_df_taxid$genus])nodelab <- tree_rooted$node.label[tree_rooted$node.label %in% tree_df_taxid$genus]nodedf <- data.frame(id=nodeids)genus_to_highlight=c("Bifidobacterium","Enterococcus","Bacteroides","Fusobacterium","Collinsella","Clostridium")nodedf <- nodedf%>% filter(nodelab %in% genus_to_highlight)nodedf$type <- as.factor(genus_to_highlight)p5 <- p4 +geom_hilight(data=nodedf, mapping=aes(node=id),extendto=8, alpha=0.3,size=0.03)p6 <- p5 + geom_hilight(data = nodedf,mapping = aes(node = id),fill = c('red','red','#009f86','#009f86','#3c5487','#4dbad5'),extendto = 8,alpha = 0.3,size = 0.03)p6
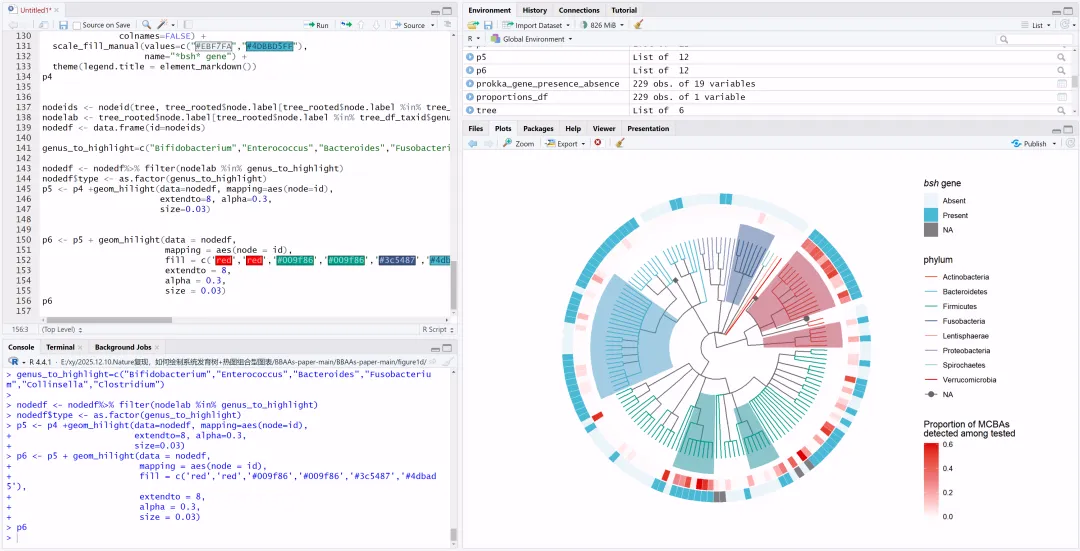
-
tree_rooted$node.label中有一些内部节点的 label 是属名(genus)。 -
nodeid():找到这些属对应的内部节点 ID。 -
genus_to_highlight:选定要高亮的几个属。 -
geom_hilight(... extendto=8):在选中的 clade 外面画一个浅色高亮区域,延伸到比较外围(extendto=8),让该属对应的所有 tip 所在的扇形区域被淡淡地罩住。
生物学解释:这几个属是文献中重点讨论的菌群,比如与胆汁酸代谢、肿瘤微环境、疾病状态高度相关,通过高亮可以让读者一眼看到这些属在树上的分布和其 MCBA/bsh 特征。
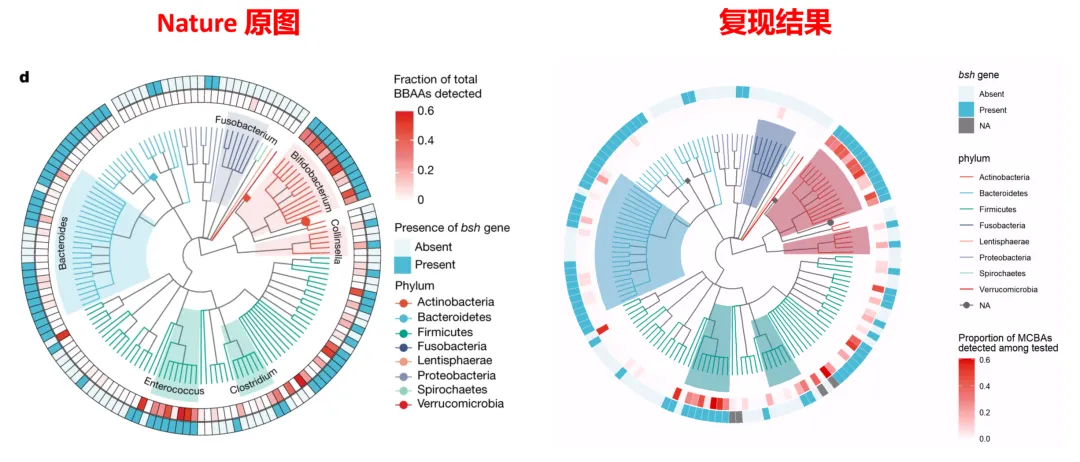
8. 总体图的生物学含义
把图整体想象成:
-
中间是彩色环形系统发育树:
-
颜色 = phylum(门)
-
每条分支 = 一个菌株
-
第一圈(靠近树的内圈 heatmap):
-
每条对应一支 tip 的条带
-
填色 = 该菌株在实验中检测到的 MCBA 产生比例(proportion)
-
越红表示越善于产生 MCBAs
-
第二圈(更外圈的窄条带):
-
两种颜色 = bsh gene 是否存在
-
表达了“基因层面功能潜力”
-
外面的淡色扇形高亮区域:
-
标出若干关键属,如 Bifidobacterium、Bacteroides、Clostridium 等
-
比较:这些属在 phylogeny 上的位置、它们的 MCBA 产生能力、以及 bsh 的携带情况。
这样一张图同时整合了:系统发育(phylogeny)+ 分类学(phylum/genus)+ 基因型(bsh presence)+ 表型(MCBA 产生比例),而且还能看出不同属之间的模式差异,是典型 CNS 图的思路。
零基础想学生信,看👇这个文章:
欢迎关注 | 华哥科研平台

往期精彩内容
亲,写的这么辛苦,记得关注、点赞、打赏哟!

