 夜雨聆风
夜雨聆风





医疗器械GMP系列解读三(文件数据+设计开发)
前言/PREFACE
新版《医疗器械生产质量管理规范》共十五章132条,将于2026年11月1日起施行。整理一下,一起学习。未来几年,只有真正建立起科学、严谨、可持续的质量管理体系的企业,才能在激烈的市场竞争和严格的监管环境中脱颖而出。
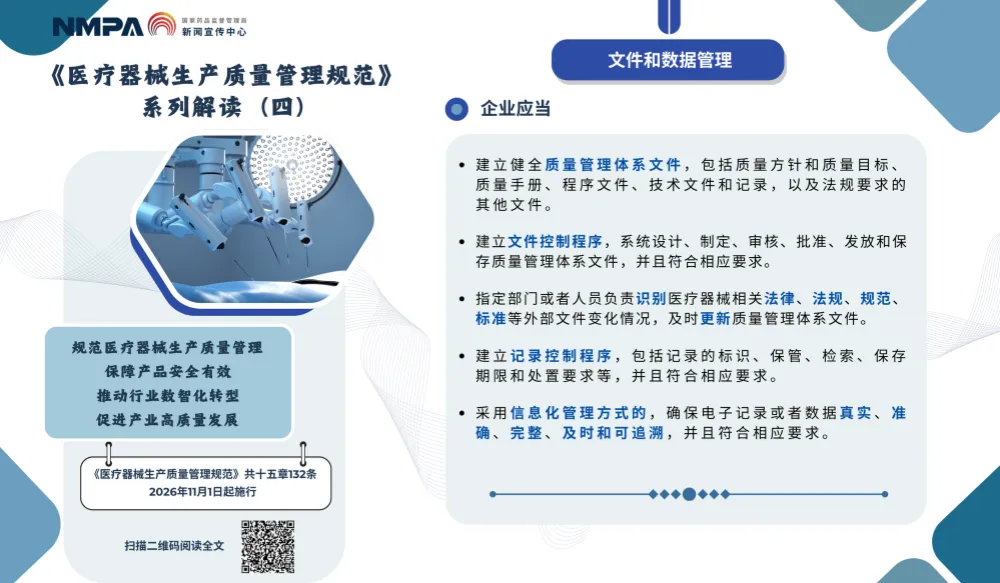
以上图片来源于国家药监局的系列解读四,聚焦于新版《医疗器械生产质量管理规范》中关于“文件和数据管理”的核心要求,旨在确保企业质量管理体系的系统性、可追溯性和合规性。
文件和数据管理
1.建立健全质量管理体系文件
➜ 包括:质量方针、目标、手册、程序文件、技术资料、记录等。
👉 就像“企业的操作指南”,每个岗位都得按流程办事。
2.建立文件控制程序
➜ 文件要经过设计、制定、审核、批准、发放、保存等流程,不能随意修改或使用旧版本。
👉 比如:某工艺文件更新后,必须收回旧版,防止工人误用过期文件。
3.及时识别并更新外部法规文件
➜ 指定的责任部门或责任人要关注国家药监局发布的最新法规、标准变化,并同步更新内部文件。
👉 如即将于2026年11月实施的GB/T 16294-2025《医药工业洁净室(区)沉降菌的测试方法》,要及时修订相关文件。
4. 建立记录控制程序
➜ 记录要有标识(如编号)、保管方式、检索方法、保存期限(通常不少于2年)和销毁规定。
👉 比如:检验记录要标明日期、检验人、设备编号,方便日后查证。
5.信息化管理下的电子记录要求
➜ 使用MES、LIMS、ERP等系统时,电子记录必须做到“四真一追”:真实、准确、完整、及时、可追溯。
👉 例如:某批产品检测不合格,系统能查到谁做的、什么时间做的、用了哪个仪器,全过程清晰可见。

以上图片来源于国家药监局的系列解读五,聚焦于强化“设计开发”环节的要求。这是新版规范中非常关键的一环,旨在从源头上保障医疗器械的安全性、有效性与可追溯性。
强化“设计开发”环节要求
1. 建立设计开发控制程序
👉 不允许“边做边改”或“凭经验搞研发”。
👉 必须有清晰的流程图和责任分工。
👉 每个阶段都有明确的输入、输出、评审标准。
📌 示例:某血糖仪开发项目分为5个阶段:需求分析 → 设计 → 样机试制 → 验证 → 确认,每个阶段完成后需提交报告并审批。
2. 根据产品特性进行设计开发策划
👉 针对不同产品的风险等级(一类、二类、三类)和复杂程度,明确各阶段的具体活动。
👉 高风险产品(如心脏起搏器)需要更严格的策划。
👉 包括:确定设计输入(用户需求、法规要求)、输出(图纸、软件代码)、验证方式等。
👉 可参考ISO 13485、IEC 60601等国际标准。
💡 实务:在策划书中列出“关键设计参数”及其验收标准,避免后期争议。
3. 开展设计开发到生产的转换活动
👉 将设计成果转化为可重复、可控的生产工艺,并确保相关规程得到验证并适用于商业化生产。
👉 这是“研发转量产”的关键一步,必须完成:工艺文件编制、设备调试、人员培训、小批量试产。
👉 所有转换活动需经过评审与批准,不能直接跳过试产就大规模生产。
⚠️ 风险点:某企业直接按样机图纸投产,导致批量产品尺寸偏差,属于严重缺陷。
4. 对设计开发进行验证
👉 通过试验、模拟等方式,确认设计输出满足设计输入要求。
👉 “验证”= 是否符合设计要求(内部一致性)。
👉 方法包括:实验室测试、计算验证与模拟分析、原型测试、对比验证等。
👉 必须保留记录:验证方案、报告、结论、参与人员签字。
🧩 示例:人工关节设计中,用疲劳试验机测试其寿命是否达到50万次以上。
5. 对设计开发进行确认
👉 通过临床评价或其他方式,确认产品满足预期用途和用户需求。
👉 “确认”= 是否符合实际使用场景(外部适用性)。
👉 第二类器械可采用同品种比对、文献分析等方式,对于第三类器械,通常需要开展临床试验。
👉 确认结果必须作为放行上市的重要依据。
📊 数据支持:某新型手术机器人需完成不少于30例临床试验,证明其操作精度优于现有产品。
6. 识别并评估设计变更的影响
👉 对任何设计变更进行识别、风险评估、验证/确认,并在实施前获得批准。
👉 变更不仅是“改图纸”,还包括材料替换、功能调整、软件升级等。
👉 需评估对安全性、有效性、合规性的影响。
👉 重大变更可能触发重新注册或临床评价。
🔄 示例:某监护仪更换显示屏供应商,虽看似简单,但需评估显示色温是否影响医生判断,必要时需重新验证。
7. 建立设计开发文档
👉 保存所有设计开发过程中的记录,确保历史过程可追溯。
👉 文档包括:需求文档、设计图纸、验证报告、变更记录、评审会议纪要等。
👉 建议使用电子化系统(如PLM)统一管理。
👉 审核时可随时调取完整链条。
🗂️ 推荐结构:【设计开发档案】:项目立项书、设计输入清单、设计输出文件、验证报告、确认报告、变更记录
往期回顾:

你的点赞是我最大的动力
