 夜雨聆风
夜雨聆风





实验新手必读(二):PCR设计宝典、软件应用、操作细节…..
特别福利:关注 “解螺旋” 微信公众号,回复关键词“9月”可索取2016年9月资源包:PCR实验宝典。
作者:解螺旋.子非鱼
如需转载请注明来源:解螺旋·医生科研助手
遥想当年,小鱼初遇PCR时,原以为可以轻松的搞定,谁知PCR却是一个不折不扣的大坑。各种假阳性、假阴性结果齐飞,非特异性扩增条带一如狗皮膏药一般挥之不去,在寻找各种原因无果后,小鱼愤怒的捏碎了电泳胶。然而痛定思痛后,小鱼发现PCR之所以能成为科研路上的拦路虎,首先,引物设计可算是一大功臣。


当然首推的经典之法就是让Premier5携手oligo,共同设计PCR引物。

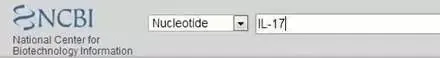
1\以小鼠的IL-17为例,进入NCBI界面,选择Nucleotide后,输入IL-17,点击search。
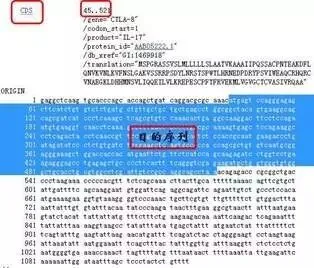
然后选择人类IL-17的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,选定目的序列。
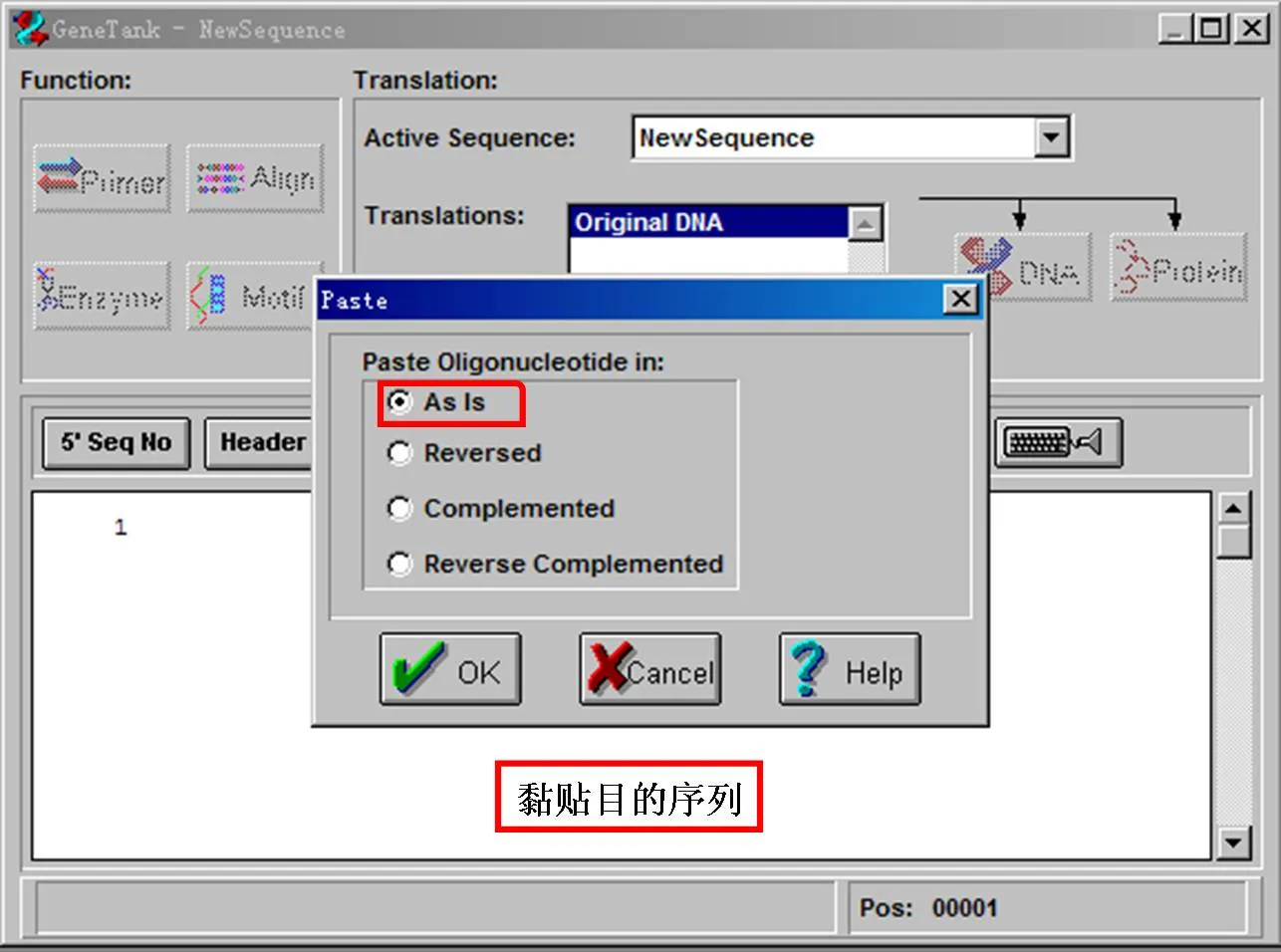
2、打开Primer Premier5软件,点击菜单栏File|New|DNA Sequence,在出现的对话框里黏贴目的序列,并在paste对话框里选择As Is,点击OK。
3、点击Primer,弹出菜单后点击search按钮。
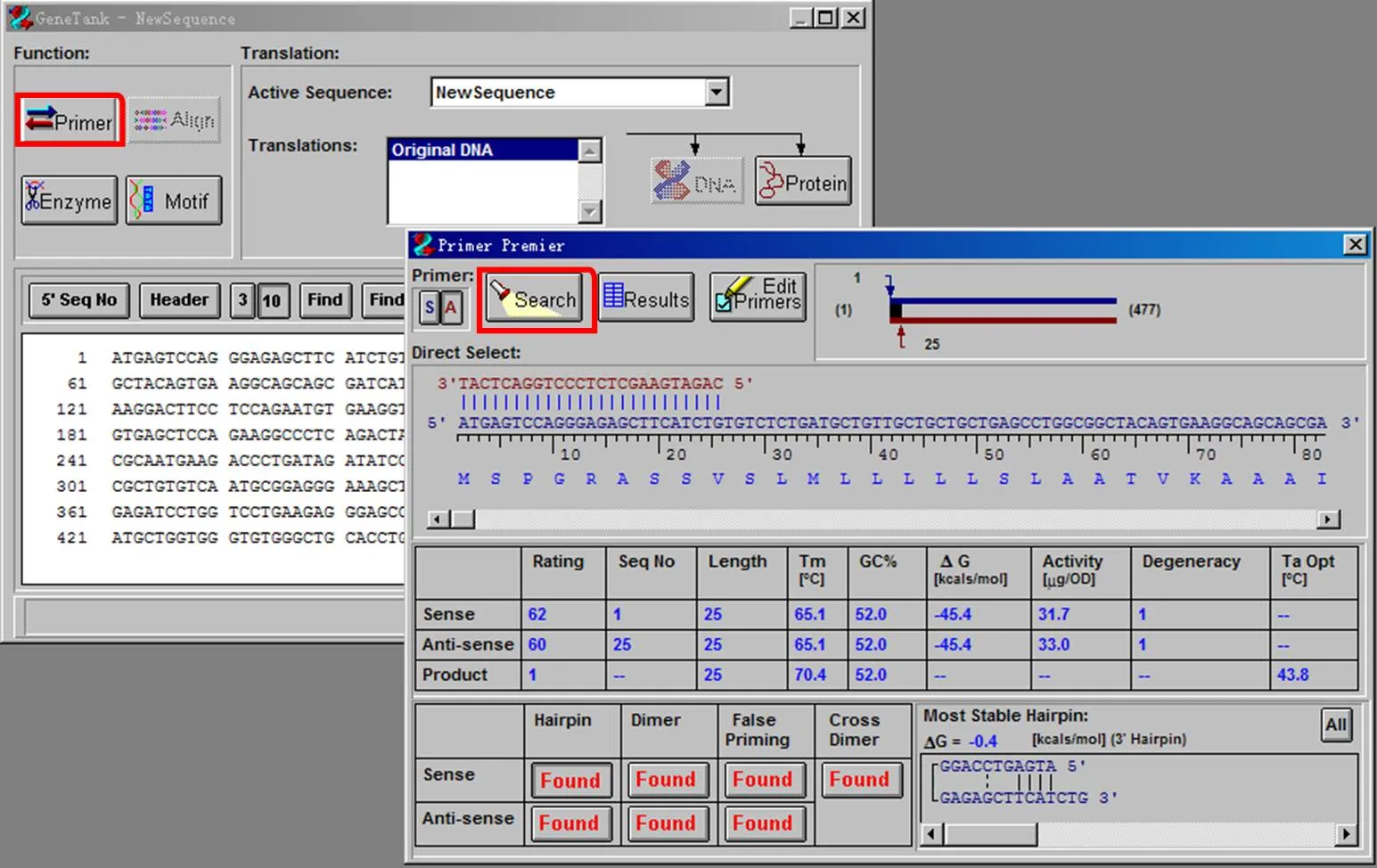
在弹出的Search Criteria中,选择PCR primers,Pairs 参数,并选择所需
PCR产物长度,点击OK。
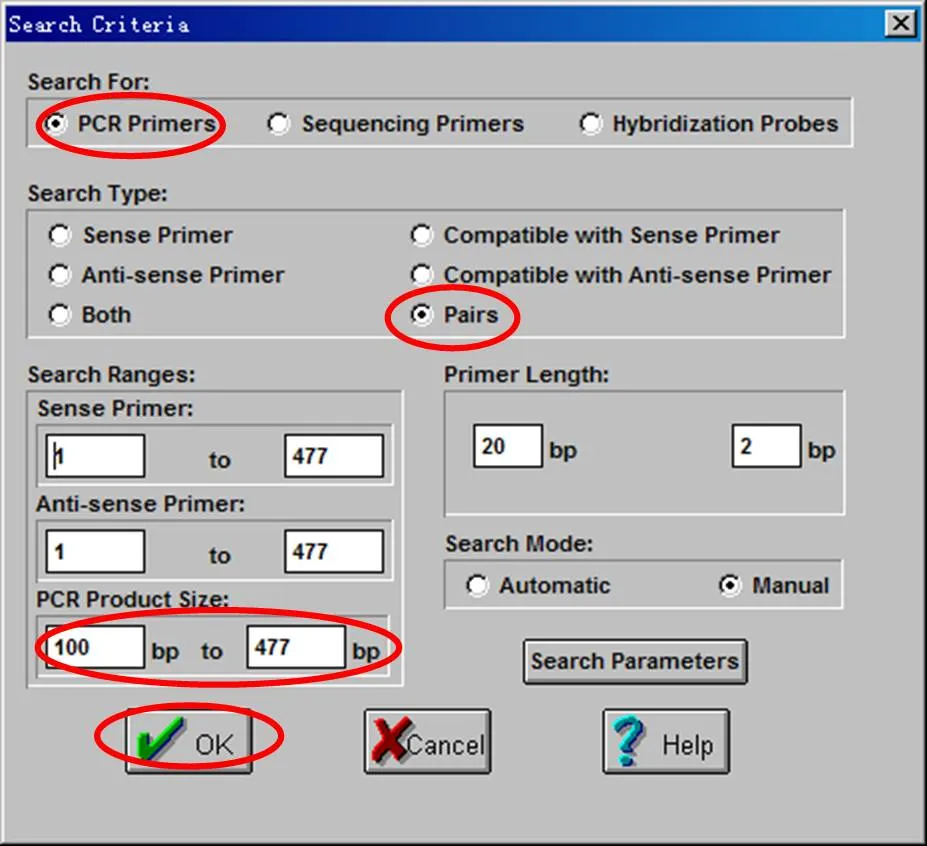
在弹出的search progress菜单中,点击OK。
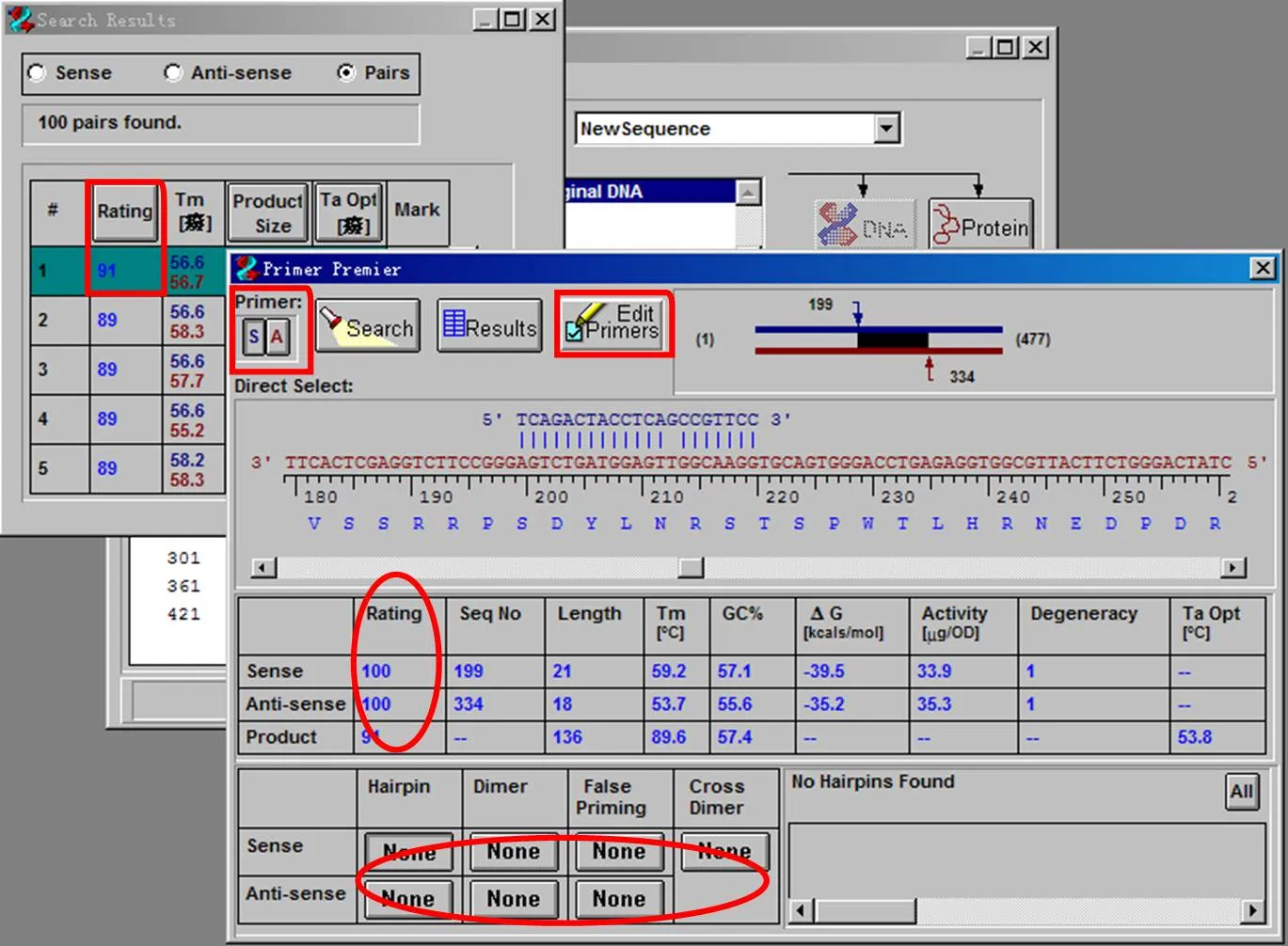
4、在搜索出的结果列表里选择Rating值高,bug较少的引物#1,在Primer Premier中选择S(正义链)/A(反义链),点击edit primer,将所得引物改成为Rating值为100,无bug的引物,即无发夹(Hairpin)、无二聚体(Dimer)、无错配(False Priming)和交叉配对(Cross Dimer)的引物。
5.点击菜单栏中Edit∣Copy∣Sense Primer/Anti-sense Primer,即可得到设计的引物。
Anti –sense Primer: 5′ GGTGGTCCATCTTTCCCT 3′
6.接下来,就要用Oligo软件验证评估Premier5软件所设计的引物,打开Oligo界面如下:
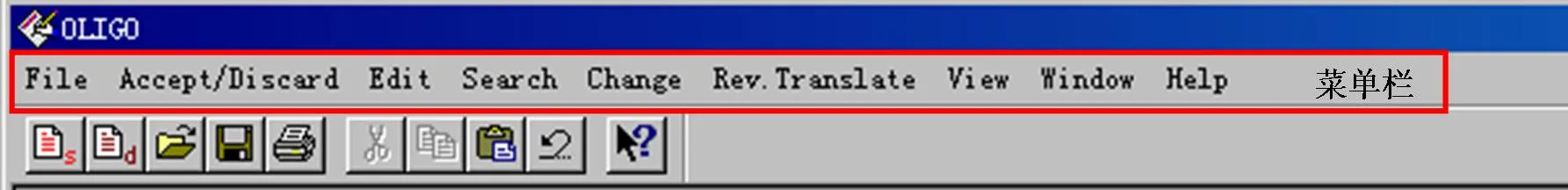
单击菜单栏里File∣New Sequence可打开以下窗口,并将设计好的上游引物黏贴在空白框里。
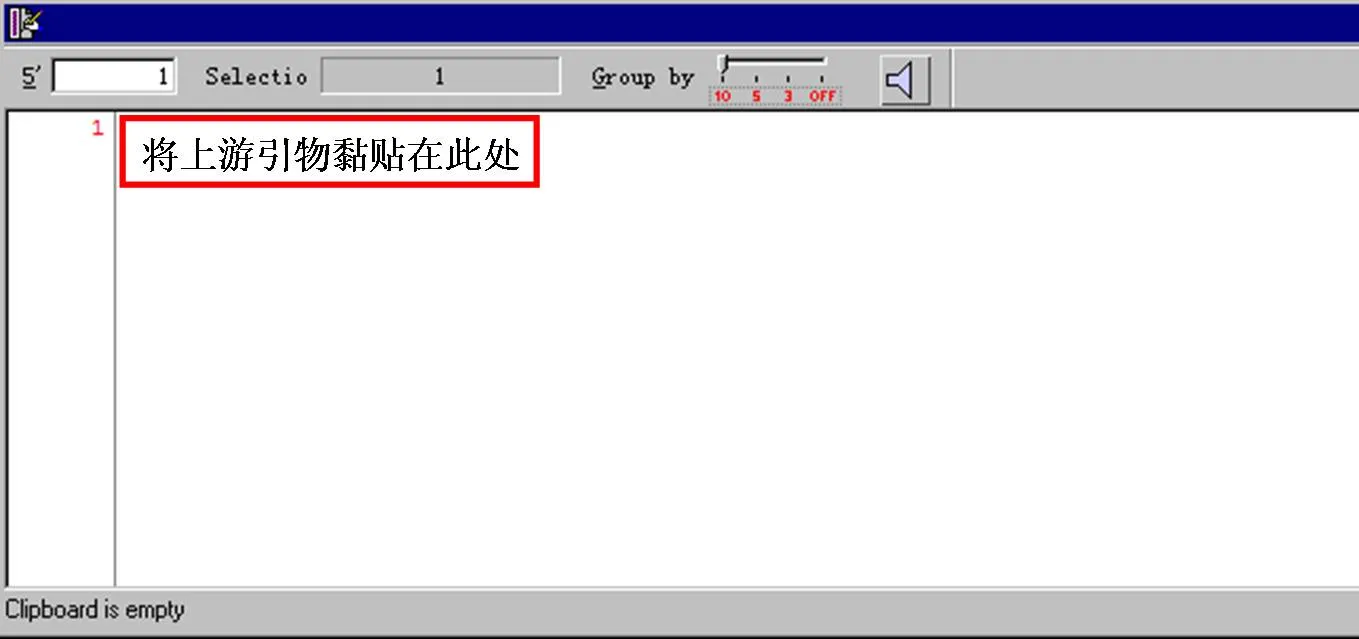
7.点击菜单栏里Accept/Discard中的Accept,则会出现Tm、△G和Frq三个窗口, △G值反应了序列与模板的结合强度,最好引物的△G值在5’端和中间值比较高,而在3’端相对较低。
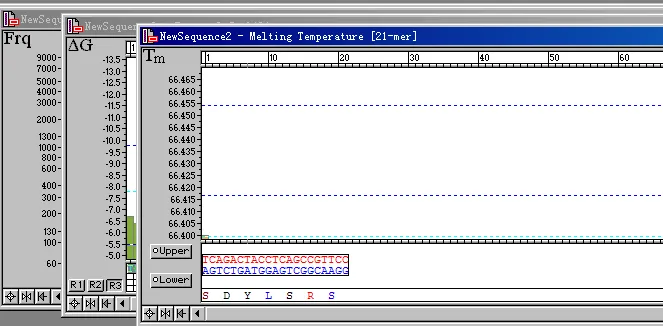
8、点击菜单栏里的Select中的Upper Primer将当前引物设置为上游引物。同时点击菜单栏里的Edit中的“Lower Primer”命令,在Edit Lower窗口中输入下游引物的序列。
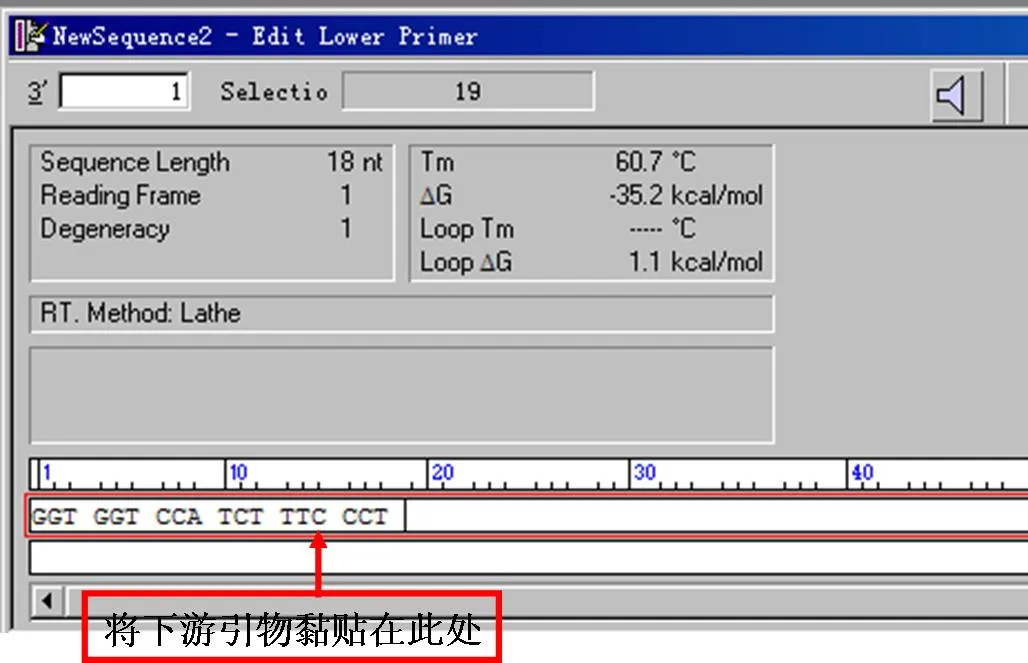
9、最后,在菜单栏中Analyze完成引物△G值、Tm值、引物二聚体和发夹结构的分析后,可将引物序列送至公司进行合成即可。
然而,此法虽好,但是下载软件总归是件麻烦事儿!有木有更简单粗暴的设计引物的方法呢?答案是YES!有!这3款简单实用的在线PCR引物设计软件可以说是懒癌晚期患者的福音。


No.1 NCBI的Primer-Blast
1、打开以下网址:http://www.ncbi.nlm.nih.gov/tools/primer-blast/ 进入Primer-Blast界面,开始引物的设计。
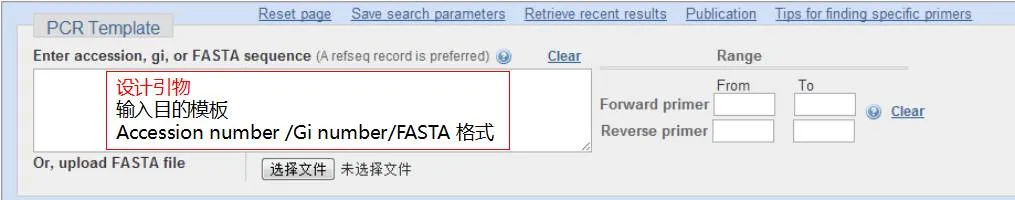
2、验证引物好坏
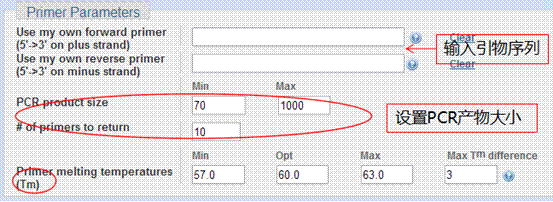
3、外显子内含子(Exon/intron)
如果设计的引物是跨内含子和外显子的交界处,可在此处设置成为Primer must span an exon-exon junction。外显子的匹配碱基数和内含子的长度等设置一般选择默认。
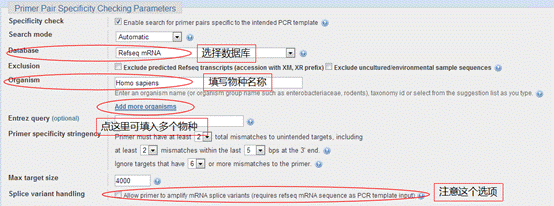
4、特异性(Specificity check)
在specificity check区,选择设计引物或验证引物时的目标数据库和物种。这一步是比较重要的。其中,数据库中的RefSeq mRNA和Genome (reference assemblies from selected organisms)是经过专家注释的数据,可得出更准确的结果。
5、最后建议用oligo6或primer5验证引物自身的特性:发夹结构、引物自身二聚体、错配和引物间二聚体,△G的绝对值。
No.2 Primer3 Plus
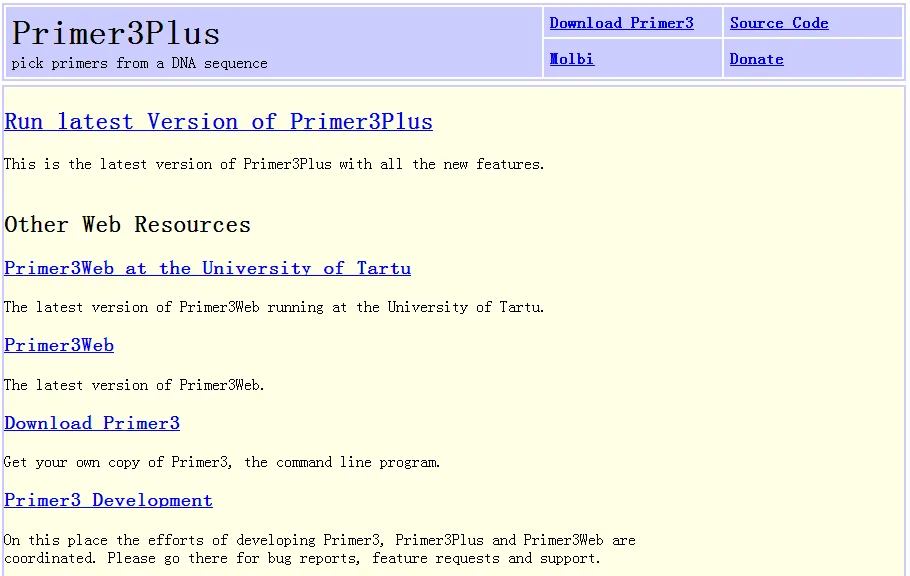
点击Run latest Version of Primer3Plus进入了设计引物的主页面。
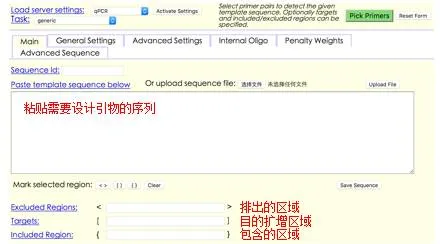
2、在General Settings中,依照引物设计原则,设置产物长度(<=400bp,最好100-150bp)、GC%(最好为45%-55%)、引物长度(最好为22-24bp)、TM值(58-60℃,最好60℃)。
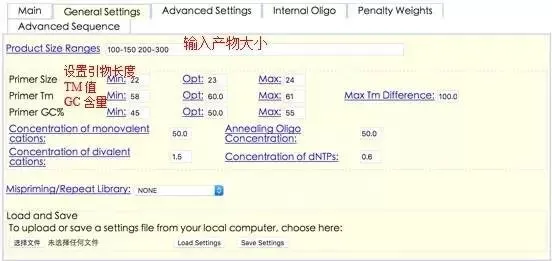
3、点击右上放绿色按钮 ,可获得若干对引物,并按照5’到3’的顺序,包含了引物和产物的一些基本参数。此时,应尽量选择G、C结尾的引物进行合成。
,可获得若干对引物,并按照5’到3’的顺序,包含了引物和产物的一些基本参数。此时,应尽量选择G、C结尾的引物进行合成。
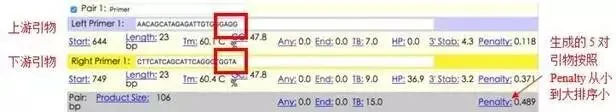
4、设计好引物后,仍需用NCBI blast 引物验证,点击http://blast.ncbi.nlm.nih.gov/Blast.cgi可出现以下界面。
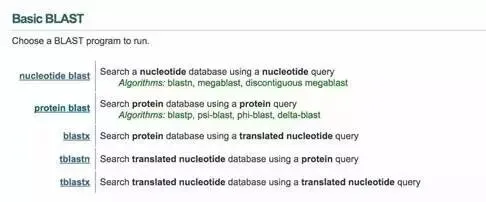
5、点击Basic BLAST中的nucleotide blast选项,在下面框中,按照5’-3’顺序,分别输入上游引物和下游引物,上下游引物之间间隔20个以上n。
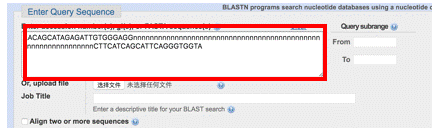
6、选择物种数据库,如Human,Mouse或者Others(如果是非人和非小鼠物种)。
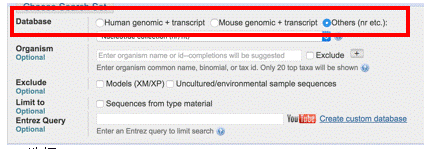
7、选择Somewhat similar sequences(blastn)

8、点击Algorithm parameters, 在Expect threshold 输入1000,在Word Size(读长)输入7,便可按照7个一组核苷酸序列放到GeneBank中进行比对。
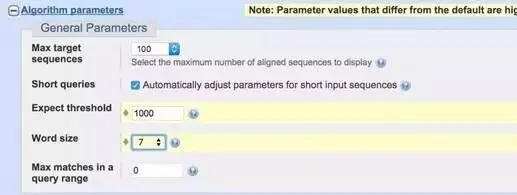
9、点击BLAST进行比对得出结果。颜色代表得分,选择引物的原则是:列表中大部分是目的基因,说明引物特异性高;但是可能物种不同,说明该基因在不同物种中保守。

No.3 Primerbank
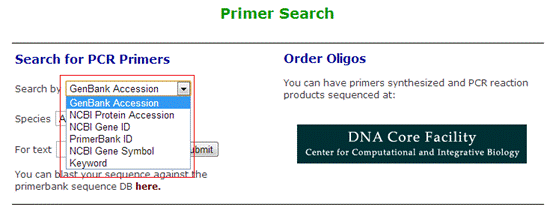
1、点击该网址:http://pga.mgh.harvard.edu/primerbank/,进入搜索界面。搜索类型这里,可以根据GenBank accession、NCBI protein accession、NCBI gene ID、primerbank ID、NCBI gene symbol以及keyword六个选项进行搜索。
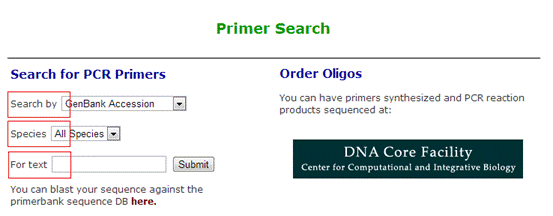
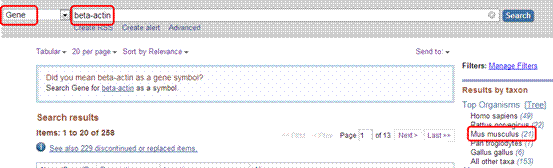
2、以小鼠的beta-actin为例。首先到NCBI上找出β-actin的gene ID,如图,选择gene,输入beta-actin,并点击Results by taxon中的Mus musculus,可得到以下结果。
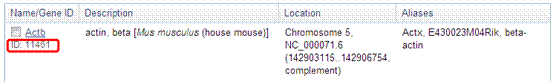
小鼠beta-actin的NCBI gene ID是11461,而actb则是小鼠beta-actin的NCBI gene symbol。
3、将Gene ID:11461输入到primerbank点击submit,可得到以下结果。
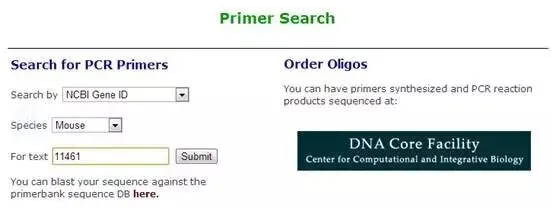
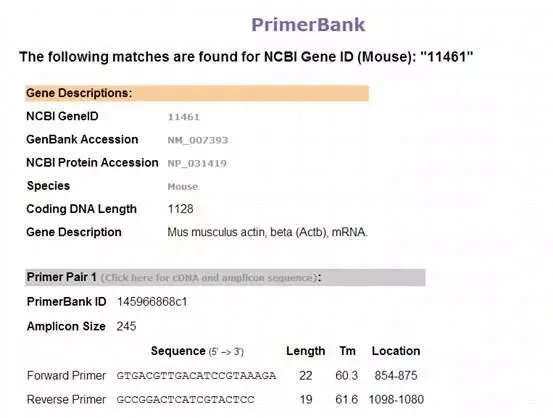
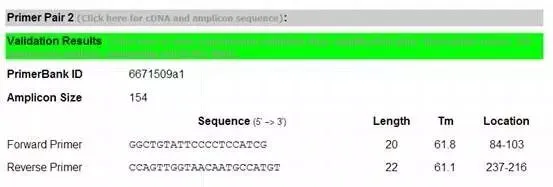
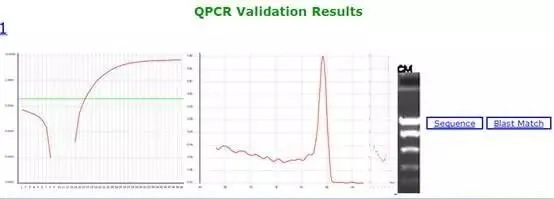
4、往下拉动滚动条,还可看到经过验证的引物,点击Validation Results,即可看到溶解曲线、电泳结果等信息。


1、最好使用一次性进口tip头,以避免液体残留;同时,tip头不要长时间暴露于空气中,避免气溶胶的污染。
2、加样时,tip头要伸入PCR反应管的液面下一点,然后将液体缓缓打入液面下,至恰好打出一个小气泡为至;加样过程最好在冰块上操作;移液枪用完之后要归到最大计量的位置,防止久而久之弹簧失去弹性,而导致加样量的不准确。
3、配制反应体系时,若反应物为冻结制品,需在融解后上下颠倒轻轻均匀混合;加入试剂之前,把它混匀一下,以免放置时间长了浓度不均;按照液体体积由大到小的顺序将反应物加入到PCR管中;引物和模板和体系所加的比例要合适,模板过量反而会抑制体系的反应。
4、 操作多份样品时,可先制备反应混合液,即将dNTP、缓冲液、引物和酶混合好后分装,这样即可避免污染,又可增加反应的精确度。
5、避免反应液飞溅,打开反应管时为避免此种情况,开盖前稍离心;PCR产物的电泳检测时间一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚至消失。


可是有时即便小伙伴们将上述实验细节处理的妥妥当当的,依然等不来奇迹发生的时候,各种非特异性条带、拖带等糟心的实验结果纷纷出来给大家心中添堵。在此,小鱼归纳了几类PCR常见的问题,并将其相应的解决方法分享给大家。

Q1:PCR产物出现假阳性(即空白对照出现目的扩增产物)
Answer:
1)引物设计不合适:扩增序列与非目的扩增序列有同源性,PCR也可扩增出非靶序列的序列;靶序列太短或引物太短,可导致假阳性。此时需重新设计引物。
2)为了避免靶序列受到整个基因组或大片段的交叉污染,操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外;除了酶及不能耐高温的物质外,所有试剂及器材应高压消毒,所有离心管及加样枪头等均应一次性使用;必要时,加样前反应管和试剂均用紫外线照射,以破坏存在的核酸;
Q2:PCR产物中出现假阴性或无扩增产物(即阳性对照中有条带,而样品则无条带)
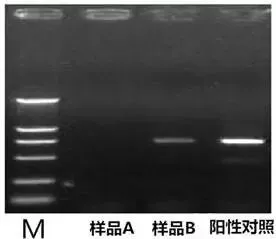
Answer:
1)条带放置时间过久,核酸被降解,最好在48h内进行电泳检测。
2)DNA模板纯度低,如含有杂蛋白质或Taq酶抑制剂,此时可对DNA进行再次纯化或重新用优质试剂盒提取DNA; DNA浓度太低时,可以加大模板量;对具有二级结构DNA使用较好的聚合酶;提取DNA时,避免吸入酚类试剂。
3)对设计不合理的引物进行重新设计合成;引物应高浓度小量分装保存,防止多次冻融而降解失效;检测引物OD值并进行电泳检测以确保两条引物浓度一致。
4)酶失活时,可更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而导致假阴性。
5)PCR反应条件:提高变性/退火温度;适当增加循环次数。如果反应系统中污染了蛋白酶及核算酶,则在未加Taq酶以前,将反应体系95℃ 加热 5~10 分钟。
6)Mg2+浓度过低可影响PCR扩增产量甚至使PCR扩增失败,而Mg2+浓度过高会降低PCR的特异性,因而可适当提高Mg2+浓度。
Q3:非特异性条带扩增或者条带出现拖尾现象
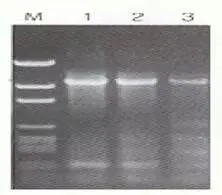
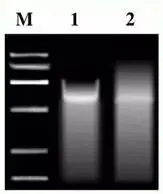
Answer:
1)当引物特异性差或引物形成二聚体时,可重新设计引物或者使用巣式PCR。
2)若模板或引物浓度过高,可适当降低模板或引物浓度,如质粒DNA<50ng,基因组DNA<200ng。
3)酶量过多,则适当减少酶量;酶质量差,可调换另一来源的酶。
4)Mg2+浓度偏高,则降低镁离子浓度。
5)退火温度偏低,适当提高退火温度或使用二阶段温度法(94℃变性,65℃左右退火与延伸)
6)循环次数过多,不仅会降低扩增效率,且会使错误掺入率增加,因此需要减少循环次数。
7)电泳体系有问题:凝胶中缓冲液和电泳缓冲液浓度相差太大;凝胶没有凝固好;琼脂糖质量差。
Answer:
四种策略:1)巣式PCR(Nest-PCR)可增加稀有靶序列的灵敏度;降低了扩增多个靶位点的可能性;提高PCR特异性。

2)递减PCR(Touch Down PCR):前几个循环使用严谨的退火条件提高特异性;循环设在比估算的Tm高大约5的退火温度下开始,然后每个循环降低1-2,直到退火温度低于Tm 5 。适合用于AFLP、DNA指纹分析等。
3)热启动PCR:抑制一种基本成分延迟DNA合成,直到PCR仪到达变性温度。如在冰上配制PCR反应液以抑制Taq酶活性,后将其置于预热的PCR仪中。
最后,小鱼把这首魔性的PCR之歌送给大家,与君共勉!

61:非编码RNA类型及功能汇总,吐血推荐!
62:一文读懂 | 与自噬相关的mTOR信号通号
63:干货 | Oligo设计引物,就是这么简单
64:跟着13分文章学作图,等着收获SCI吧(origin8教程)
65:干货 | 磷酸化抗体使用必杀技
66:Discussion写作模板:从3分、5分到10分
67:一文包会:Web of science数据库应用宝典
68:读图 | qPCR那些奇奇怪怪的曲线都代表啥?
69:MicroRNA,如何实现从零基础到10分的跨越
70:ELISA实验操作中值得关注的细节大盘点
回复SCI、国自然、信号通路、CNS、实验工具、统计查看相应专栏文章!
想与同行同领域探讨学术,交流感想吗?扫描加入分领域专业社区吧!

投稿邮箱: tougao@helixlife.com.cn
合作微信:helixlife6

