 夜雨聆风
夜雨聆风





硬核升级|宏基因组分析云流程16款分析工具+6大功能数据库全新上线,重塑宏基因组分析
 点击蓝字↑↑↑“微生态”,轻松关注不迷路
点击蓝字↑↑↑“微生态”,轻松关注不迷路

一. 六大功能数据库——权威扩容+优化,功能注释更精准
数据库是宏基因组功能解析的核心支撑,本次升级全面扩容数据库资源,新增6大权威数据库并优化现有数据库,覆盖多研究方向,让基因功能注释更贴合研究需求、结果更具说服力。
-
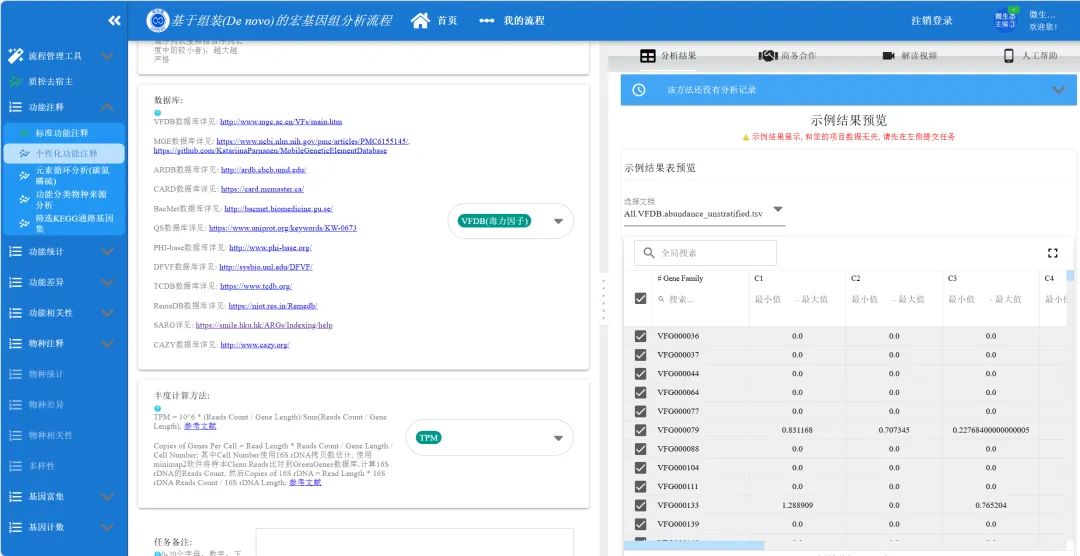
SARG(v3.2)功能注释数据库:新增专属抗生素抗性基因注释数据库,精准识别样本中抗生素抗性基因,为耐药微生物研究、环境抗性风险评估提供核心数据支撑。
-
KEGG 通路数据库:同步更新KEGG通路数据库,升级后成果体现在KEGG通路丰度表与通路图中,过往已交付流程可通过“更新流程”功能,一键同步最新KEGG通路丰度表,无需重复分析,保障历史数据同步升级
-
PHI-base(病原-宿主互作数据库):收录海量经实验验证的病原-宿主互作基因,助力解析病原微生物感染机制、宿主免疫响应规律,适配动植物病原、人体致病菌等相关研究。
-
DFVF(真菌毒力因子数据库):聚焦真菌致病性研究,全面收录真菌毒力相关基因与蛋白,快速挖掘真菌致病核心靶点,为真菌病害防控、病原溯源提供关键线索。
-
TCDB(转运蛋白数据库):权威微生物转运蛋白注释资源,覆盖各类物质转运相关基因,清晰解析微生物营养吸收、代谢产物分泌、耐药物质转运等核心生理过程。
-
RemeDB(污染物降解基因数据库):环境微生物修复研究专属数据库,收录各类有机、重金属污染物降解相关基因,助力高效降解菌群筛选、污染环境修复机制研究。
-
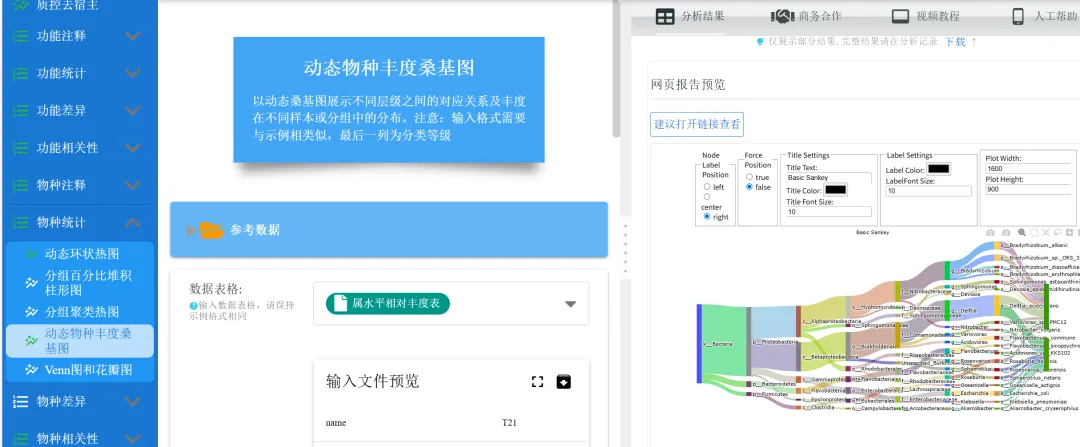
物种丰度桑基图:以动态桑基图展示不同层级之间的对应关系及丰度在不同样本或分组中的分布。
-
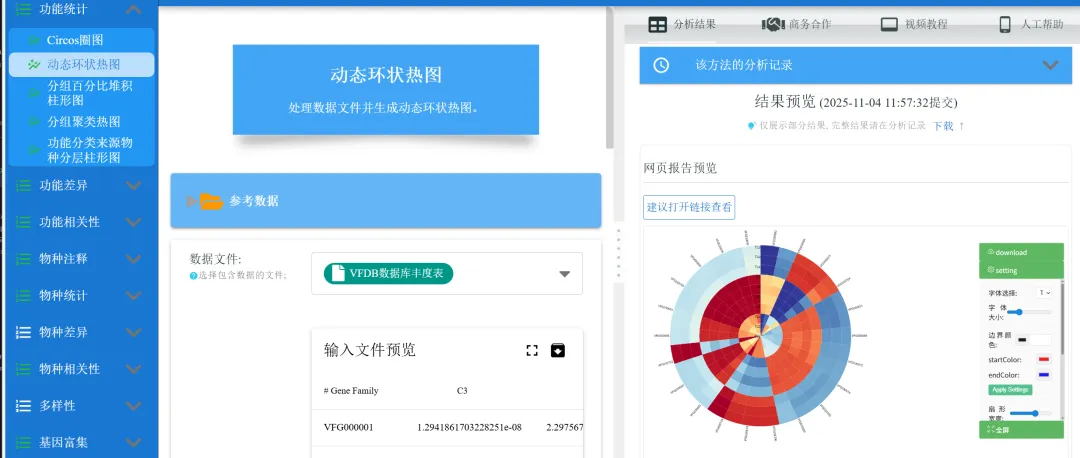
环状热图:突破传统平面局限,环形布局叠加物种/样本信息,凸显核心差异菌群。
-
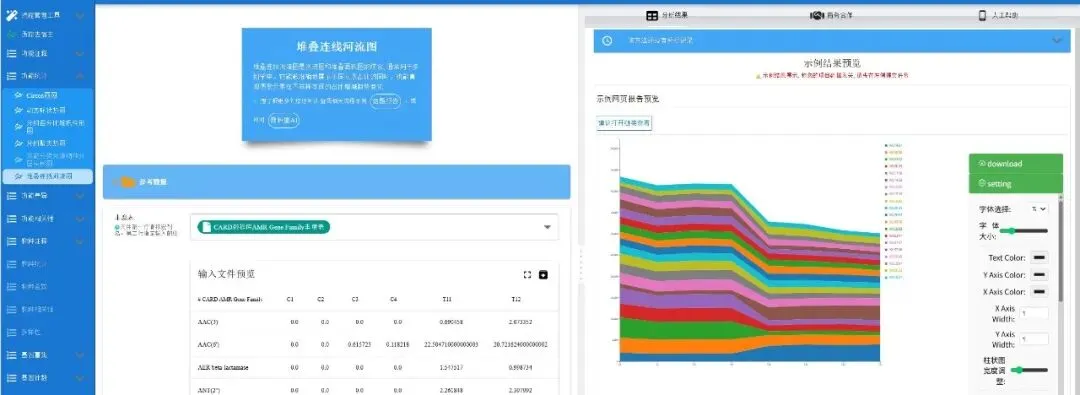
堆叠连线河流图:堆叠连线河流图是河流图和堆叠面积图的结合, 通常用于多组学中。它能够准确地展示不同元素占比的同时,也能直观感受元素在不同样本间的占比增减趋势变化。
-
两组/多组比较横向柱形图:对于两组, 综合计算和可视化分组均值, 差异比较p值, 差异置信区间, Combine Effect Size, Log2FC等指标。对于多组, 计算和可视化分组均值, 差异比较p值。
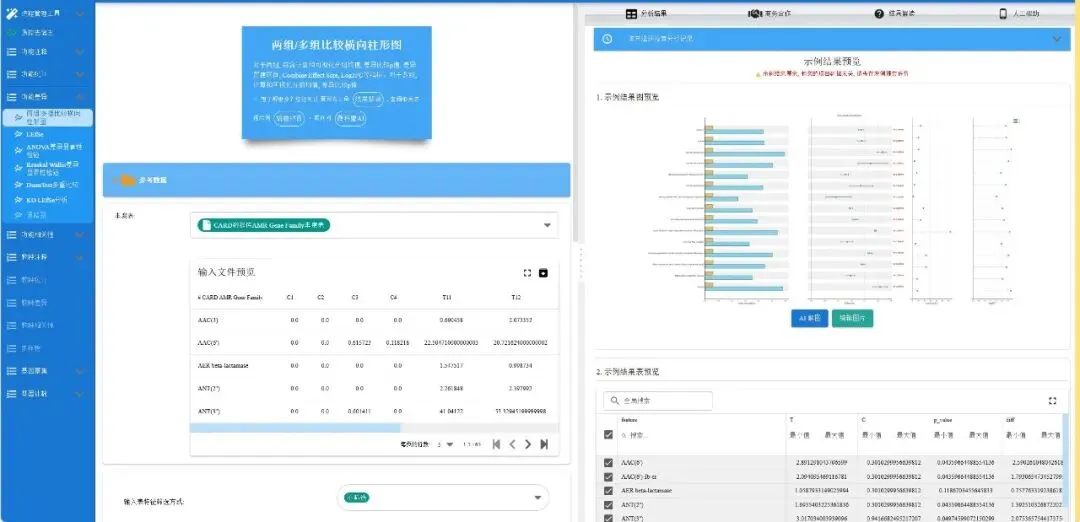
-
普鲁克分析[Procrustes Analysis]:基于R vegan包, 多组学关联分析,以揭示两组数据的一致性。
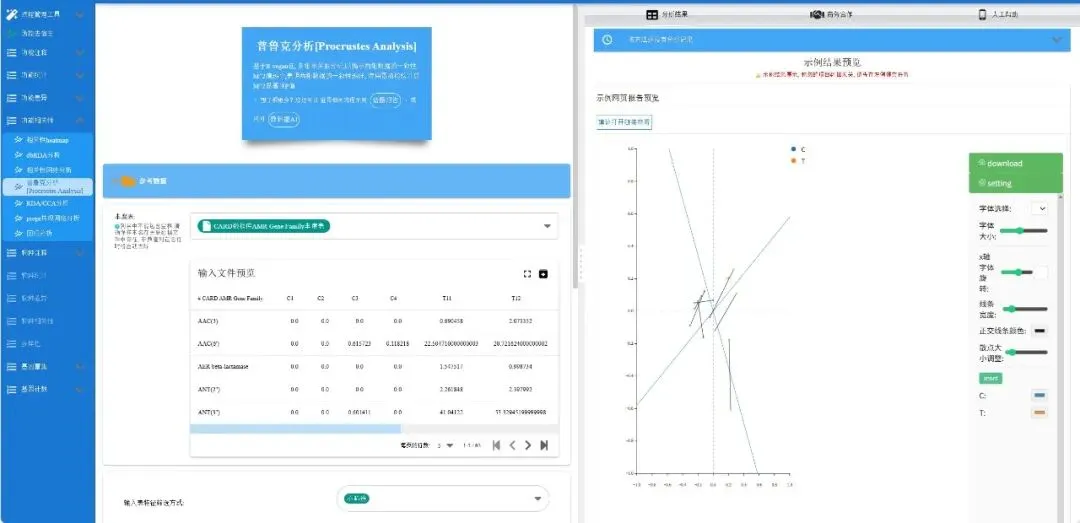
-
propr共现网络分析:通过propr包中的相关性分析不同微生物种群或基因之间的相关性,揭示它们在环境或人体中的共现模式。
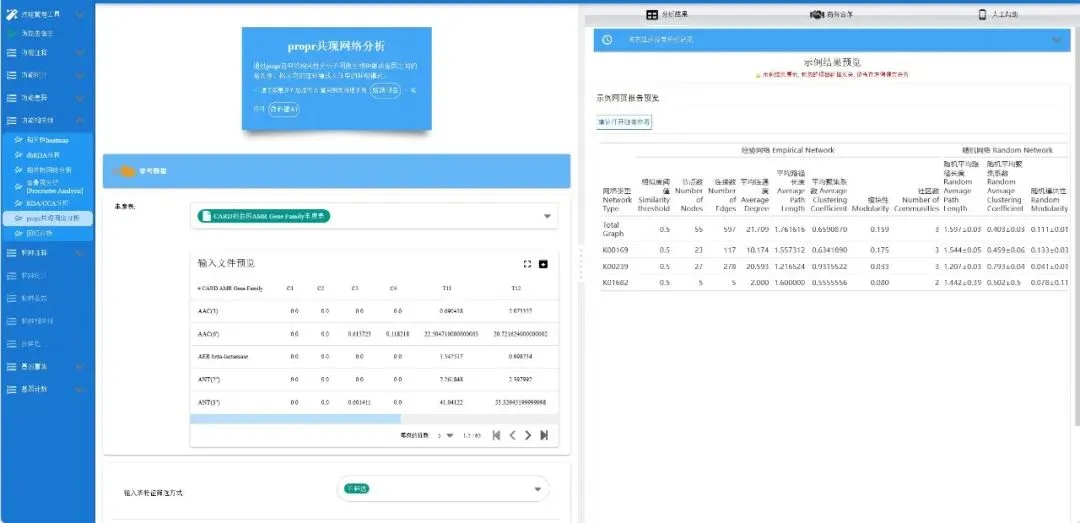
-
物种分类热树图:以热树图形式展示丰度表中物种分类关系。
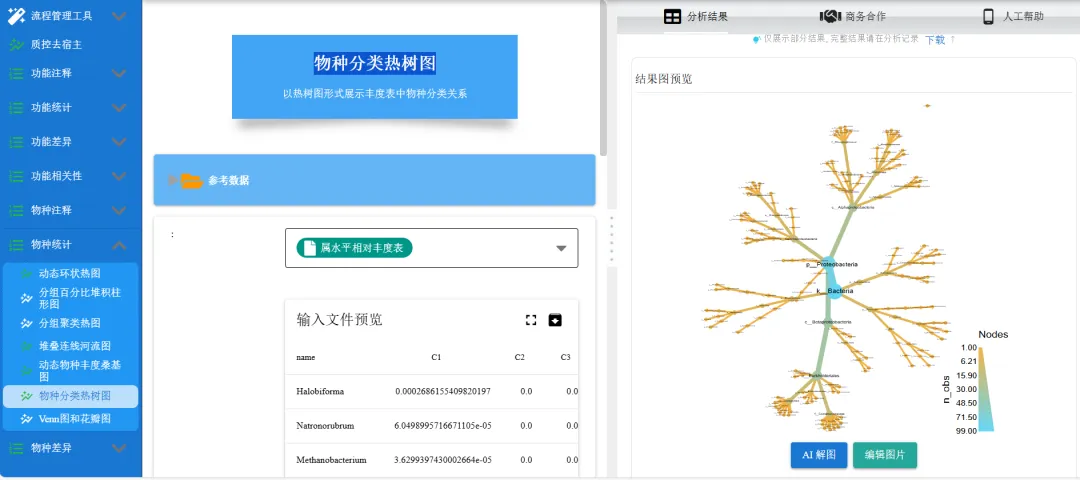
-
共同丰度组CAG聚类分析:CAG聚类分析及可视化,根据丰度表对特征进行聚类为CAG,探索CAG内(丰度箱线图)、CAG之间(网络图)、CAG与环境因子之间(热图)的关系。
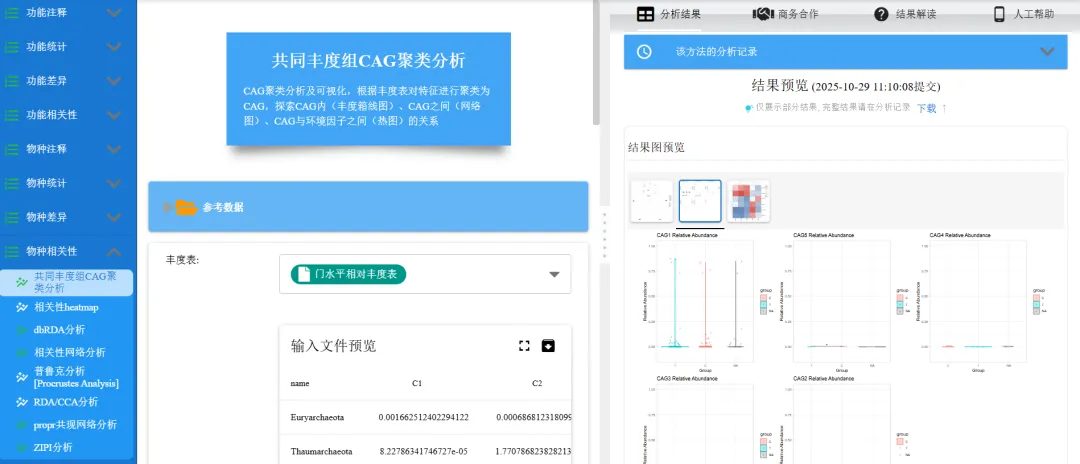
-
dbRDA(距离矩阵冗余分析):一种多变量分析技术,用于探索环境因子对样本间变异的影响。它结合了经典的冗余分析(RDA)和距离矩阵方法,能够处理非线性关系和复杂的生态数据。
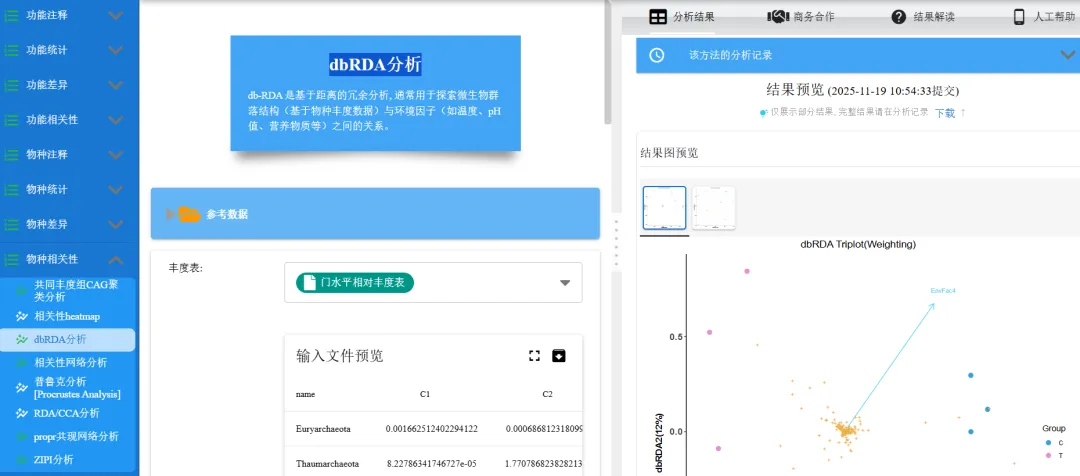
-
ZIPI分析:寻找微生物关联网络中的关键物种
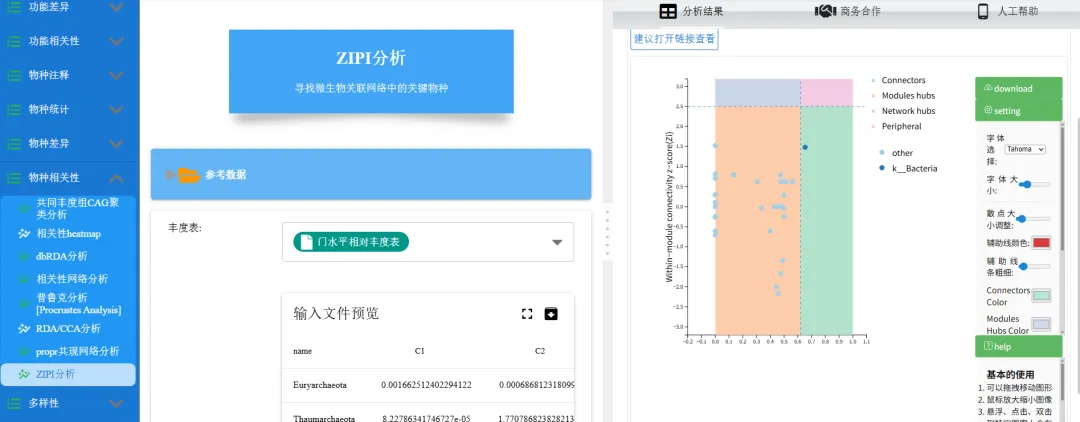
-
肠型分析:基于样品间的Jensen-Shannon距离,利用围绕中心点划分算法(PAM)进行聚类,最佳分类数目通过Calinski-Harabasz (CH)指数确定。
-
PCA主成分分析:将多features的高维丰度表,降为二维,方便比较组间整体差异。
-
Qiime2 β多样性分析:将多features的高维丰度表,降为二维,方便比较组间整体差异。
-
Simper分析:一种用于生态学数据分析的方法,常用于探究群落结构之间的差异,并确定哪些物种对这些差异贡献最大。
-
tSNE:通过将高维数据集中每个数据点映射到二维或者是三维地图上的位置来实现数据可视化,属于非线性的降维方式。
-
UMAP:一种基于非线性降维的可视化方式,将高维数据映射到二维或三维空间,并保持数据之间的相对距离和结构,从而使得聚类、异质性和样本间的差异更为明显。 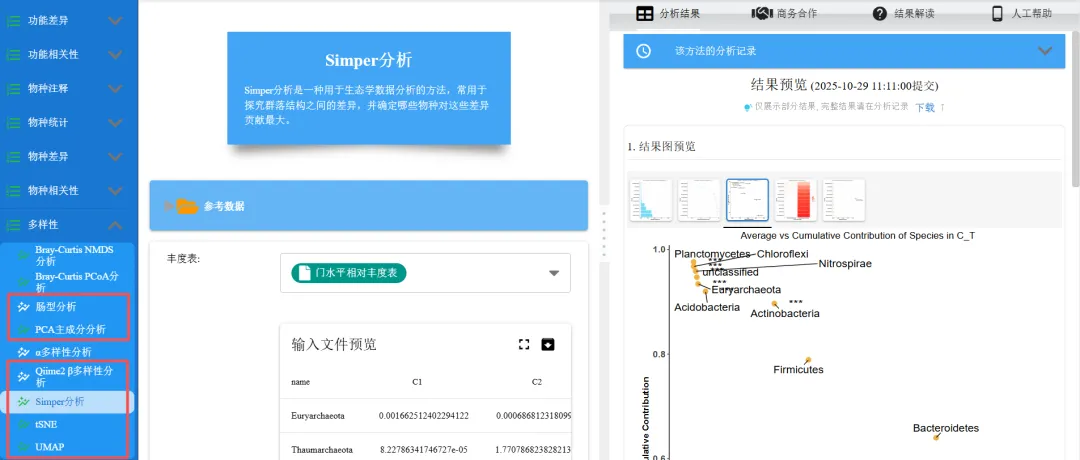
本次更新升级,实现「功能注释精准化、统计分析深度化、可视化展示多元化、多样性解析全面化」的四大突破。全程云端运行,无需本地配置环境、无需编写复杂代码,一键提交任务即可自动生成标准化分析报告与高清可编辑图表,直接适配SCI论文发表要求。
未来,我们将持续迭代优化,为全球宏基因组科研工作者提供更专业、更易用的数据分析工具。如您在使用过程中有任何疑问或建议,欢迎随时联系微科盟组学老师。已添加微科盟组学老师的请直接联系已添加的组学老师,如果您没有添加过微科盟组学老师,请添加多组学老师27,请勿重复添加。

微文推荐阅读
微科盟智能科研平台—”微科盟AI”重磅上线!科研综述、实验方案设计、期刊查询…一个AI全搞定!
微科盟智能科研平台——”微科盟AI”体验网址:https://wekemo.bioincloud.tech/litoverse-home。
或者点击微信小程序微科盟体验
 获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系多组学老师27,无需重复添加。
获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系多组学老师27,无需重复添加。