 夜雨聆风
夜雨聆风从2015年开始生物信息培训,目前已经走过10个年头,累计学员超过30万。从2021年推出《基因学苑生物信息VIP课程》以来,已有超过1000人学习过我们课程。2025年我们隆重推出《基因学苑VIP课程(2025版)》,本套课程是我们10年集大成制作,也是之前课程的全面升级版。新版本课程现已开售。
包含服务内容
1、1042集生物信息视频
2、6个月上机操作
3、赠送30万文字讲义
4、赠送课程配套PPT
5、赠送全部课程案例代码
6、微信答疑群
课程特色
1、2025最新视频
2、从零开始教学,适合小白
3、4K分辨率视频
4、加入AI辅助生物信息内容;
5、分成更多章节,结构清晰,方便碎片化时间学习
6、5年有效期;
7、包括6个月上机操作,全部案例亲自上手操作一遍;
8、包含课程配套30万字讲义,大量PPT,10000+案例代码;
9、微信答疑群;
10、可开具发票。
课程目录:
上机操作
云服务器配置为128核心,1024G内存,1024G磁盘,里面包含全部课程案例代码,登录云服务器,可有对照视频完成全部案例课程代码。且配置有国际网络专线,没有网络限制,下载速度更快。
如何购买
1、渠道一:
添加下面微信,转账付款,发送下载地址,进行课程解锁,获取账号,开发票等事宜。
手机微信扫描下方二维码

2、渠道二:
淘宝付款购买:复制下面地址到浏览器中或者使用手机淘宝扫描二维码,付款完添加上面微信。
淘宝链接:https://item.taobao.com/item.htm?spm=a21dvs.23580594.0.0.6ffb645ea5q4SL&ft=t&id=619716421433
手机淘宝扫码二维码

常见问题:
1、上机操作服务器是什么?
2、课程是否适合零基础?
本课程完全从零基础开始,包含理论讲解与案例操作,是一套系统学习生物信息的优秀资料。里面包含了生物信息基础,Linux,常用工具,测序原理,生物软件管理,R,python等基础内容,还包括RNAseq,单细胞,宏基因组,16S,人基因组变异检测等应用内容。适合零基础从头开始学习。
3、是否能只购买课程一部分?
本课程为打包销售,不单独售卖。
4、想在多台设备播放怎么办?
课程为本地加密版本,只能在一台电脑设备上播放,windows系统和苹果系统均可,不支持手机端播放,也不支持录屏。因为视频内容很大且里面有很多操作演示,不适合手机端播放。默认只支持一台设备,如果想多台设备播放,每台加1000元。
5、课程可以看多久?
课程有效期到2030年12月31日。
6、如何进行解锁?
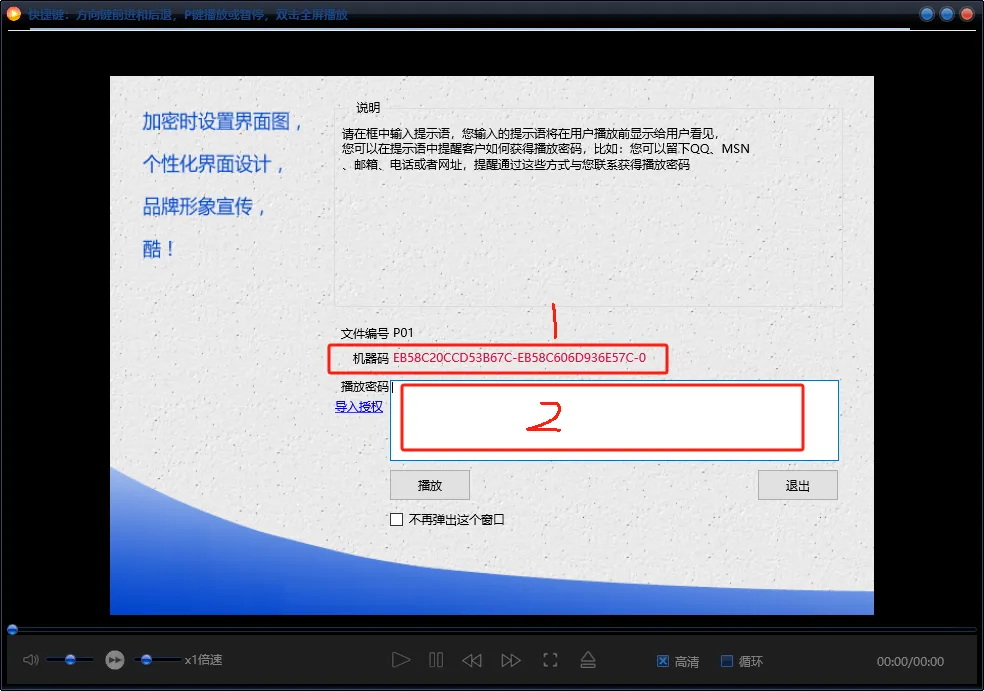
视频需要使用专用播放器进行播放,下载播放器,导入任何一个视频,将下图1所示的机器码发给我即可进行解锁,然后将解锁码填入图中2部分即可,一次解锁即可观看全部内容。
7、是否可以开发票
本教程提供发票,发票为电子发票,具体信息可添加上面微信进行咨询。
8、课程学习顺序
对于零基础用户,建议按照课程1-零基础生物信息入门,2-生物信息常用工具,3-测序原理这样的顺序学习,这部分为基础操作部分,同时配合操作。掌握基础之后后面可以按照需要的部分进行个性化针对性学习。
9、课程可以试看吗
下面地址提供课程试看部分,
链接: https://pan.baidu.com/s/19VsOzG6qcICeAH9SMXjb_A?pwd=emrj 提取码: emrj
课程详细目录
下面列出课程主要内容以及详细目录
1-零基础生物信息入门
| ||
1-课程介绍 | 2-测序行业发展历史(2005-2008) | 3-测序行业发展历史(2009-2010) |
4-测序行业发展历史(2011-2013) | 5-测序行业发展历史(2014) | 6-测序行业发展历史(2015) |
7-测序行业发展历史(2016-2017) | 8-测序行业发展历史(2018-2020) | 9-测序行业发展历史(2021-2024) |
10-生物信息应用 | 11-学生物信息如何划拉钱 | 12-为什么生物信息比较难学 |
13-什么是服务器? | 14-为什么做生物信息需要服务器? | 15-熟悉Linux操作系统 |
16-登录服务器准备工作 | 17-安装termius软件 | 18-使用termius登录服务器 |
19-termius设置 | 20-通过网页登录服务器 | 21-传输文件 |
22-使用filezilla传输数据 | 23-使用vscode登录服务器 | 24-什么是命令行操作 |
25-命令行基本操作 | 26-命令行常用快捷键 | 27-选项参数 |
28-不同系统平台目录结构 | 29-使用cd和ls操作目录 | 30-必须用tab补齐 |
31-绝对目录与相对目录 | 32-Linux系统创建目录和文件 | 33-Linux系统复制文件操作 |
34-Linux系统创建快捷方式 | 35-Linux系统移动重命名和删除文件 | 36-Linux系统查看文件 |
37-使用head查看文件 | 38-Linux数据流 | 39-管道 |
40-文件压缩 | 41-多线程压缩 | 42-打包文件 |
43-多线程打包压缩 | 44-统计文件 | 45-Linux权限 |
46-修改权限 | 47-什么是脚本 | 48-使用vim编辑脚本 |
49-vim使用案例 | 50-预测基因案例 | 51-使用echo生成脚本 |
52-Linux进程管理 | 53-Linux前后台任务切换 | 54-Linux非挂起任务 |
55-tmux不间断会话 | 56-tmux使用案例 | 57-解决Linux报错信息 |
58-Linux环境配置 | 59-给命令行添加颜色 | 60-PATH变量 |
61-什么是生物软件? | 62-生物软件如何发文章? | 63-安装生物软件几种方法 |
64-如何查找生物软件 | 65-什么是分析流程? | 66-如何编译软件 |
67-bioconda简介 | 68-不同的conda版本有什么区别? | 69-下载bioconda |
70-安装bioconda | 71-使用bioconda安装生物软件 | 72-记录软件版本 |
73-使用bioconda管理生物软件 | 74-升级软件 | 75-bioconda常见问题 |
76-虚拟环境 | 77-使用虚拟环境安装软件 | 78-管理虚拟环境 |
79-安装rnaseq分析环境 | 80-使用管理员bioconda环境 | 81-导出软件配置 |
82-普通用户安装依赖环境 | 83-bioconda管理R分析环境 | 84-创建测试账户 |
85-服务器之间直接传输数据 | 86-迁移软件 | 87-使用命令行传输数据 |
88-使用condapack迁移环境 | 89-容器技术 | 90-容器生物软件的缺点 |
91-安装docker和apptainer | 92-使用docker安装生物软件 | 93-使用apptainer安装生物软件 |
94-使用apptainer | 95-在容器中安装软件 | 96-安装deepvariant |
97-下载生物数据简介 | 98-生物数据下载方法大全 | 99-下载基因组序列 |
100-使用lftp命令下载数据 | 101-从nmdc下载数据 | 102-SRA数据库简介 |
103-下载测序数据 | 104-安装awscli | 105-使用awscli下载测序数据 |
106-使用迅雷下载数据 | 107-使用for循环批量操作 | 108-使用while循环批量操作 |
0-linux.zip |
2-生物信息常用工具
| ||
1-git和github | 2-安装和使用git | 3-使用gitbash |
4-通过终端使用git | 5-配置git | 6-如何使用git |
7-git版本控制 | 8-github简介 | 9-为什么使用github |
10-注册和使用github账号 | 11-提交github | 12-fork项目 |
13-github设置 | 14-github其他功能 | 15-连接git与github |
16-github密钥登录 | 17-服务器自动登录 | 18-vscode简介 |
19-安装和配置vscode | 20-安装插件 | 21-vscode远程登录服务器 |
22-vscode自动登录 | 23-远程打开文件 | 24-vscode快捷键 |
25-vscode中使用sftp功能 | 26-vscode中使用git | 27-什么是codespaces |
28-创建codespaces | 29-配置codespaces | 30-codespaces作为测试服务器 |
31-利用AI辅助数据分析 | 32-豆包AI | 33-使用TraeAI下载数据 |
34-安装TraeAI插件 | 35-使用TraeAI生成代码 | 36-使用TraeAI修改选项参数 |
37-使用TraeAI配置环境 | 38-使用TraeAI绘图以及名词解释 | 39-使用TraeAI咨询方案以及解决报错 |
40-使用TraeAI开发程序 | 41-编写统计fastq文件程序 | 42-使用TraeAI解释代码 |
43-利用copilot辅助生物信息分析 | 0-ai_analysis.zip |
3-测序原理
| ||
1-为什么要学习测序原理 | 2-测序技术发展历史 | 3-不同测序平台比较 |
4-自己测序还是外包 | 5-DNA测序四个步骤 | 6-提取DNA |
7-illumina测序特点 | 8-illumina测序原理 | 9-构建文库步骤 |
10-构建文库原理 | 11-测序芯片flowcell | 12-cluster |
13-read1测序 | 14-read2测序 | 15-双末端测序 |
16-illumina测序碱基识别 | 17-matepair文库 | 18-测序常见问题 |
19-fastq文件格式 | 20-phred质量值转换 | 21-Q20标准 |
22-数据质控原理 | 23-fastqc质控illumina测序数据 | 24-fastqc结果解读 |
25-illumina过滤原理 | 26-为什么要去除duplication | 27-利用fastp过滤数据 |
28-过滤结果解读 | 29-multiqc整合结果 | 30-pacbio平台简介 |
31-sequel测序系统 | 32-revio测序系统 | 33-onso测序系统 |
34-SMRT测序原理 | 35-pacbio聚合酶 | 36-哑铃壮文库 |
37-hifi测序reads | 38-pacbio测序错误来源 | 39-获取pacbio测序数据 |
40-pacbio数据质控 | 41-pacbio数据过滤 | 42-filtlong过滤pacbio数据 |
43-pacbio原始数据转换 | 44-onso测序结果质控过滤 | 45-pacbio数据处理总结 |
46-nanopore平台简介 | 47-荧光信号的优缺点 | 48-电信号的优缺点 |
49-nanopore测序发展历史 | 50-选择合适的纳米孔 | 51-nanopore测序芯片 |
52-nanopore测序原理 | 53-nanopore建库 | 54-选择合适的试剂盒 |
55-选择合适的nanopore测序仪 | 56-估算纳米孔数据产出量 | 57-国产纳米孔测序平台 |
58-碱基识别原理 | 59-nanopore测序错误来源 | 60-如何提高纳米孔测序准确性 |
61-nanopore测序优点 | 62-nanopore测序缺点 | 64-nanopore测序碱基识别 |
64-配置nanopore测序分析软件 | 65-nanopore测序数据质控 | 66-Q20plus数据质控 |
0-sequencing.zip |
4-基因组数据分析
| ||
1-为什么要拼接基因组? | 2-目前已测序基因组查询 | 3-为什么基因组难拼接 |
4-不同平台拼接比较 | 5-获取基因组大小 | 6-基因组拼接原理简介 |
7-什么是kmer | 8-为什么要使用kmer | 9-kmer估计基因组大小 |
10-二代测序拼接算法 | 11-处理重复序列 | 12-输出contig |
13-构建scaffold | 14-处理gap区域 | 15-案例文章简介 |
16-获取文章数据 | 17-批量下载测序数据 | 18-使用迅雷快速下载 |
19-下载参考基因组 | 20-使用datasets工具下载基因组 | 21-安装分析软件 |
22-sra数据转换 | 23-数据质控过滤 | 24-kmer频数统计 |
25-kmer估计基因组大小 | 26-安装配置spades软件 | 27-使用spades拼接基因组 |
28-spades输出介绍 | 29-SOAPdenovo简介 | 30-SOAPdenovo配置文件 |
31-利用SOAPdenovo拼接基因组 | 32-补洞 | 33-如何选择kmer大小 |
34-为什么kmer只能取奇数 | 35-如何判断污染序列 | 36-拼接结果统计 |
37-N50与N90 | 38-seqkit处理fasta序列 | 39-提取序列 |
40-利用quast进行结果评估 | 41-quast结果解读 | 42-安装和配置busco |
43-利用busco进行结果评估 | 44-测序数据与拼接结果比对 | 45-tablet可视化比对结果 |
46-samtools查看比对结果 | 47-pacbioCLR数据质控 | 48-pacbioCLR数据拼接 |
49-pacbioCLR结果质控 | 50-pacbioHifi数据拼接 | 51-配置nanopore测序分析软件 |
52-nanopore测序碱基识别 | 53-nanopore测序拼接方案 | 54-nanopore测序数据质控 |
55-Q20plus数据质控 | 56-nanopore基因组拼接 | 57-nanopore拼接结果评估 |
58-nanopore拼接结果纠错 | 59-比较纠错结果 | 60-基因预测原理 |
61-prodigal原核生物基因预测 | 62-glimmer3原核生物基因预测 | 63-在线网站预测基因 |
64-同源比对预测基因原理 | 65-同源比对预测基因 | 66-安装配置Augustus |
67-利用Augustus对拟南芥基因预测 | 68-基因功能注释原理 | 69-常用功能注释数据库 |
70-安装和配置eggnog | 71-下载数据库 | 72-使用eggnog进行基因功能注释 |
73-注释结果解读 | 74-在线基因功能注释 | 75-ncRNA分析原理 |
76-安装和配置rnammer | 77-rRNA分析 | 78-tRNA分析 |
79-重复序列分析原理 | 80-trf预测串联重复序列 | 81-配置repeatmasker |
82-使用repeatmasker重复序列分析 | 83-blast比对原理 | 84-配置blast比对环境 |
85-利用blast进行局部比对 | 0-assembly.zip |
5-宏基因组数据分析
| ||
1-什么是宏基因组? | 2-宏基因组研究对象 | 3-宏基因组发展历史 |
4-宏基因组研究目的 | 5-宏基因组应用 | 6-宏基因组研究反面案例 |
7-使用16S序列进行物种分类 | 8-扩增子测序优缺点 | 9-宏基因组优缺点 |
10-为什么宏基因组测序比较难? | 11-纳米空宏基因组测序 | 12-AI辅助宏基因组分析 |
13-安装bioconda | 14-安装宏基因组分析软件 | 15-配置物种分类数据库 |
16-从aws下载kraken2物种分类数据库 | 17-数据库完整性校验 | 18-自行构建数据库 |
19-安装宏基因组拼接软件 | 20-下载练习数据集 | 21-物种分类概述 |
22-物种分类原理 | 23-taxonomy物种分类数据库 | 24-安装和配置taxonkit |
25-taxonkit检索物种分类数据库 | 26-不同物种分类方法比较 | 27-不同物种分类软件比较 |
28-为什么选择kraken2 | 29-利用kraken2进行物种分类 | 30-kraken2结果解读 |
31-物种分类结果可视化 | 32-生成桑基图 | 33-为什么物种分类结果比真实更多? |
34-比对结果过滤 | 35-使用全部测序数据分析 | 36-pavian更多功能介绍 |
37-krakentools简介 | 38-提取reads拼接基因组 | 39-转换为mpa格式 |
40-合并mpa格式文件 | 41-计算丰度 | 42-统计多样性 |
43-krona绘图 | 44-二代测序物种分类 | 45-比较二代与三代测序物种分类 |
46-通过饼图比较二代测序与三代测序 | 47-去除宿主污染 | 48-利用生物信息方法去除宿主 |
49-模拟宿主污染测序 | 50-bwa比对 | 51-使用samtools去除宿主序列 |
52-去除宿主后物种分类 | 53-临床样本检测案例 | 54-人体病源微生物检测 |
55-鉴定并拼接致病菌 | 56-批量鉴定样品 | 57-安装和使用megan |
58-megan导入物种分类结果 | 59-利用megan可视化物种分类结果 | 60-调整megan可视化绘图 |
61-多组样品比较 | 62-megan分组比较 | 63-stamp简介 |
64-stamp输入文件 | 65-kraken2结果转换为stamp输入 | 66-利用stamp进行多组检测 |
67-利用stamp进行两组比较 | 68-宏基因组拼接简介 | 69-二代测序宏基因组拼接简介 |
70-三代测序宏基因组拼接简介 | 71-拼接模拟数据 | 72-随机抽样测序数据 |
73-megahit拼接 | 74-安装和使用quast | 75-拼接结果评估 |
76-重复文章:内容解读 | 77-重复文章:安装软件和下载数据 | 78-重复文章:拼接二代测序宏基因组 |
79-重复文章:统计拼接结果 | 80-重复文章:拼接三代测序宏基因组 | 81-重复文章:先物种鉴定还是先拼接? |
82-重复文章:三代纳米孔宏基因组拼接 | 83-纳米孔测序直接拼接细菌完成图 | 84-二代与三代宏基因组测序结果比较 |
85-Q20+试剂宏基因组拼接 | 86-宏基因组基因预测 | 87-cd-hit基因去冗余 |
88-将核酸翻译成氨基酸 | 89-宏基因组功能注释原理 | 90-配置基因功能注释环境 |
91-宏基因组基因功能注释 | 92-salmon定量分析 | 93-什么是binnning? |
94-binning软件 | 95-安装和配置metawrap | 96-配置metawrap数据库 |
97-metawrap数据质控过滤 | 98-metawrap组装和分箱 | 99-分箱结果评估 |
100-宏基因组分析总结 | 0-meta.zip |
6-16S数据分析
| ||
1-扩增子测序简介 | 2-扩增子与宏基因组测序比较 | 3-扩增子测序分类 |
4-18S与ITS测序 | 5-如何选择扩增引物 | 6-不同测序平台比较 |
7-OTU与ASV | 8-HMP与EMP计划 | 9-alpha多样性 |
10-beta多样性 | 11-多样性指标 | 12-常见术语 |
13-扩增子分析软件 | 14-分析数据库 | 15-安装qiime2 |
16-使用docker安装qiime2 | 17-下载数据库 | 18-metadata |
19-探索16S序列 | 20-qiime2简介 | 21-qiime2选项参数 |
22-qiime2分析流程 | 23-导入数据 | 24-查看qza格式 |
25-导入fastq格式 | 26-manifest导入 | 27-拆分样品 |
28-拆分完统计 | 29-质控过滤 | 30-生成FeatureTable |
31-beta多样性计算 | 32-beta多样性结果解读 | 33-alpha多样性计算 |
34-绘制多样性稀释曲线 | 35-物种分类鉴定 | 36-物种分类结果可视化 |
37-qiime2总结 | 38-sabre拆分barcode | 39-dada2简介 |
40-dada2导入数据 | 41-dada2数据质控 | 42-降噪去重复 |
43-dada2生成FeatureTable | 44-dada2物种分类鉴定 | 45-dada2结果评估 |
46-dada2绘制系统发育树 | 47-dada2练习案例 | 48-phyloseq简介 |
49-phyloseq结果可视化 | 50-16S功能分析 | 0-amplicon.zip |
7-人基因组数据分析
| ||
1-人基因组分析简介 | 2-人基因组研究应用 | 3-wgs人全基因组测序技术 |
4-wes全外显子测序技术 | 5-trs目标区域捕获测序技术 | 6-参考序列对找突变的影响 |
7-生殖细胞与体细胞突变 | 8-为什么不用拼接的方法找突变? | 9-人基因组变异检测要求 |
10-安装人基因组分析软件 | 11-安装deepvariant | 12-人基因组参考序列简介 |
13-不同版本基因组比较 | 14-如何选择合适的人基因组参考序列? | 15-从aws下载人基因组分析参考序列 |
16-下载注释数据库 | 17-瓶中基因组计划 | 18-下载测序数据 |
19-测序数据与参考序列比对 | 20-建立索引 | 21-bwa比对 |
22-bwa-mem2比对 | 23-长读长测序比对 | 24-多重比对问题 |
25-sam文件格式 | 26-sam文件flag值 | 27-samtools简介 |
28-bam排序建索引 | 29-统计bam文件 | 30-按bed区间统计 |
31-tview可视化比对结果 | 32-GATK简介 | 33-GATK缺点 |
34-GATK最佳实践 | 35-数据质控过滤 | 36-GATK分析bam文件处理 |
37-利用GATK检测SNP | 38-GATK结果过滤 | 39-Deepvariant简介 |
40-利用Deepvariant检测SNP | 41-wes与trs变异检测 | 42-vcf格式简介 |
43-vcf重要指标 | 44-vcf统计 | 45-利用bcftools处理vcf文件 |
46-变异结果注释原理 | 47-纳米孔测序SV检测 | 48-利用cuteSV检测SV |
49-基因突变可视化工具IGV | 50-使用IGV可视化变异数据 | 0-human.zip |
8-RNAseq数据分析
| ||
1-RNAseq简介 | 2-RNAseq10年综述 | 3-比较DNA测序与RNA测序 |
4-RNAseq常见概念 | 5-为什么要做RNAseq | 6-原核与真核RNAseq |
7-有参于无参RNAseq | 8-bulk与单细胞RNAseq | 9-全长转录组 |
10-链特异性转录组 | 11-需要多少样本 | 12-需要多少数据量 |
13-比对基因组还是基因集 | 14-基因水平还是转录本水平定量 | 15-RNAseq定量方法 |
16-不同定量方法比较 | 17-富集mRNA | 18-RNAseq建库 |
19-spikein内参 | 20-选择合适测序平台 | 21-安装分析软件 |
22-下载参考序列和gtf文件 | 23-案例文章 | 24-下载文章数据 |
25-sra数据转换 | 26-数据质控 | 27-下载人基因组比对索引 |
28-建立star比对索引 | 29-STAR比对 | 30-AI生成批量比对脚本 |
31-featureCounts计数 | 32-处理表达矩阵 | 33-非比对方法生成表达矩阵 |
34-案例文章简介 | 35-导入数据 | 36-生成DESeq2对象 |
37-数据对象探索 | 38-比对数据统计绘图 | 39-多维数据探索 |
40-使用热图展示样品关系 | 41-使用pca展示样品关系 | 42-差异表达计算 |
43-如何筛选差异表达基因 | 44-差异表达基因可视化 | 45-使用ggplot2绘制火山图 |
46-ggvolcano绘制火山图 | 47-案例练习1 | 48-案例练习2 |
49-使用edgeR分析 | 50-使用edgeR筛选差异表达基因 | 51-注释原理 |
52-差异表达基因注释 | 53-注释练习案例 | 54-GO富集 |
55-GO富集结果解读以及可视化 | 56-KEGG注释与富集分析 | 57-下载非模式生物注释库 |
58-非模式生物功能注释 | 59-GSEA富集分析原理 | 60-配置GSEA分析环境 |
61-利用GSEAbase进行GSEA分析 | 62-利用fgsba进行GSEA分析 | 0-rnaseq.zip |
9-单细胞数据分析
| ||
1-为什么要做单细胞测序 | 2-单细胞测序发展历史 | 3-bulk与单细胞比较 |
4-单细胞研究计划 | 5-单细胞研究应用 | 6-单细胞捕获与分选 |
7-不同分选平台比较 | 8-10xgenomics简介 | 9-GEM与barcode |
10-barcode与umi | 11-构建文库 | 12-为什么选择10xgenomics |
13-单细胞测序常见问题 | 14-下载cellranger | 15-安装单细胞分析软件 |
16-下载参考序列 | 17-自建索引 | 18-获取练习数据 |
19-单细胞测序数据简介 | 20-利用cellranger生成表达矩阵 | 21-cellranger输出结果 |
22-cellranger分析结果解读 | 23-利用STARsolo生成表达矩阵 | 24-LoupeBrowser导入数据 |
25-LoupeBrowser参看单细胞结果 | 26-LoupeBrowser注释 | 27-LoupeBrowser差异表达基因分析 |
28-LoupeBrowser重新分析 | 29-使用R分析单细胞数据 | 30-Seurat简介 |
31-安装Seurat | 32-Seurat读入数据 | 33-单细胞数据探索 |
34-数据质控与过滤 | 35-数据标准化 | 36-根据差异表达进行聚类 |
37-数据降维 | 38-LoupeBrowser可视化Seurat结果 | 39-寻找标记基因 |
40-标记基因可视化 | 41-LoupeBrowser可视化标记基因 | 42-根据标记基因注释单细胞亚群 |
43-单细胞亚群鉴定原理 | 44-利用CellMarker数据鉴定细胞 | 45-对Seurat结果进行鉴定 |
46-自动化鉴定细胞类型 | 47-安装和配置singlerR | 48-下载注释数据库 |
49-singleR自动化注释 | 50-空间转录组简介 | 51-空间转录组数据分析 |
52-空间转录组结果解读 | 0-singlecell.zip |
10-生物信息分析平台搭建
| ||
1-什么是IT基础设施? | 2-什么是服务器 | 3-做生物信息为什么要使用服务器? |
4-需要多少计算资源 | 5-与IT行业区别 | 6-什么是虚拟化技术? |
7-虚拟化解决方案 | 8-开源虚拟化方案 | 9-超融合架构 |
10-什么是数据中心? | 11-共享生物云计算 | 12-服务器类型 |
13-如何选购CPU | 14-如何选购内存 | 15-如何选购硬盘 |
16-其他硬件 | 17-购买云服务器 | 18-选择debian还是redhat? |
19-redhat替代版本 | 20-带外管理 | 21-远程配置服务器 |
22-下载操作系统文件 | 23-配置虚拟机 | 24-使用虚拟机安装Linux |
25-Linux初始化设置 | 26-购买测试服务器 | 27-服务器如何连接外网 |
28-使用frp进行内网穿透 | 29-配置sudo权限 | 30-磁盘管理 |
31-raid级别 | 32-lvm逻辑卷 | 33-格式化硬盘 |
34-设置硬盘默认挂载 | 35-创建逻辑卷 | 36-磁盘限额 |
37-Linux环境配置 | 38-更改yum镜像 | 39-添加本地yum源 |
40-添加epel源 | 41-使用yum配置环境 | 42-防火墙管理 |
43-linux远程桌面 | 44-云服务器配置远程桌面 | 45-用户管理 |
46-修改账号默认配置 | 47-自动管理账户脚本 | 48-安装生物软件 |
49-配置数据库 | 50-配置R语言环境 | 51-安装R扩展包 |
52-容器技术 | 53-使用apptainer部署软件 | 54-迁移软件 |
55-ubuntu系统配置 | 56-使用apt安装生物软件 | 0-biostation.zip |
11-R语言入门
| ||
1-课程介绍 | 2-安装R和Rstudio(windows) | 3-安装R和Rstudio(macos) |
4-开始使用R | 5-rstudio设置 | 6-获取在线免费R分析环境 |
7-获取练习数据集 | 8-R语言的三种运行方式 | 9-R语言函数 |
10-获取帮助 | 11-测试计算机算力 | 12-提示警告和错误 |
13-常用快捷键 | 14-什么是R包? | 15-如何选择R包 |
16-安装R包 | 17-修改镜像 | 18-install.packages函数详解 |
19-其余R包管理函数 | 20-获取R包帮助信息 | 21-bioconductor项目 |
22-github包安装 | 23-R项目 | 24-结构化数据与非结构化数据 |
25-读入数据准备工作 | 26-点击鼠标读入文件 | 27-read系列函数读入数据 |
28-将结果写入文件 | 29-读写excel文件 | 30-读写R内置数据格式 |
31-读入纸质版表格 | 32-数据类型 | 33-向量 |
34-如何生成向量 | 35-创建字符串和逻辑值向量 | 36-向量索引 |
37-逻辑值索引 | 38-向量排序与筛选 | 39-修改向量值 |
40-向量统计 | 41-向量绘制散点图 | 42-向量绘制直方图 |
43-向量绘制条形图 | 44-向量绘制饼图 | 45-因子 |
46-计算频数 | 47-卡方检验 | 48-利用频数表绘制文字云图 |
49-矩阵 | 50-矩阵的索引 | 51-计算相关性 |
52-绘制相关性图 | 53-绘制热图 | 54-分组条形图 |
55-数据框 | 56-数据框索引 | 57-R实现vlookup |
58-数据框绘制箱线图 | 59-数据框绘制小提琴图 | 60-二维数据计算 |
61-列表 | 62-列表绘制维恩图 | 63-缺失数据 |
64-如何处理缺失值 | 65-时间序列 | 66-使用xts包处理时间序列 |
67-不同数据类型转换 | 68-二维数据排序 | 69-筛选数据 |
70-随机抽样 | 71-利用R来斗地主 | 72-数据汇总 |
73-数据探索 | 74-tidyverse | 75-修改R配置文件 |
76-tibble | 77-管道 | 78-cheatsheet |
79-tibble数据处理 | 80-dplyr处理行列 | 81-增加行列 |
82-排序去重复 | 83-筛选 | 84-分组汇总 |
85-集合运算 | 86-抽样和处理缺失值 | 87-处理字符串 |
88-整洁数据 | 89-长数据与宽数据 | 90-宽数据转换为长数据 |
91-长数据转换为宽数据 | 92-合并和拆分数据 | 93-获取真实分析数据 |
94-R虚拟环境 | 95-R语言24个高效操作技能 | 96-AI辅助R语言学习 |
97-为Rstudio添加AI功能 | 98-在Rstudio中使用AI会话生成代码 | 99-新一代R语言集成开发环境Positron |
100-R语言数据推荐 | 0-rbase.zip |
12-R语言科研绘图
| ||
1-课程介绍 | 2-安装R和Rstudio(windows) | 3-安装R和Rstudio(macos) |
4-获取在线免费R分析环境 | 5-R绘图系统 | 6-r绘图网站 |
7-R基础绘图 | 8-par绘图函数 | 9-R绘图设备 |
10-export包 | 11-mfrow绘图布局 | 12-layout绘图布局 |
13-legend图例 | 14-颜色 | 15-RColorBrewer |
16-散点图 | 17-散点图实例 | 18-增强散点图 |
19-克利夫兰点图 | 20-火山图 | 21-折线图 |
22-直方图 | 23-组合直方图 | 24-条形图 |
25-分组和堆叠条形图 | 26-箱线图 | 27-小提琴图 |
28-韦恩图 | 29-热图 | 30-文字云图 |
31-ggplot2图形语法 | 32-data数据 | 33-mapping映射 |
34-geom几何图形 | 35-ggsave保存图形 | 36-patchwork绘图布局 |
37-数据可视化案例 | 38-scale修改颜色 | 39-scale修改坐标 |
40-修改透明度与点线 | 41-Statistics统计变化 | 42-Coordinate坐标系 |
43-Layer图层 | 44-叠加图层绘制分组小提琴图 | 45-Facet分面 |
46-Theme主题 | 47-更新R和R包 | 48-theme更改背景 |
49-theme更改坐标轴 | 50-theme更改图例 | 51-通过theme修改绘图案例 |
52-ggplot2修改字体 | 53-geom_text添加注释 | 54-annotation添加注释 |
55-scales包修改标签 | 56-为条形图添加标签 | 57-添加误差条 |
58-plotly交互式绘图 | 59-ggplot2绘制散点图 | 60-ggplot2绘制火山图 |
61-ggplot2绘制增强散点图 | 62-ggplot2绘制克利夫兰点图 | 63-ggplot2绘制kegg点图 |
64-ggplot2绘制高密度散点图 | 65-ggplot2绘制气泡图 | 66-ggplot2绘制直方图 |
67-ggplot2绘制折线图和面积图 | 68-ggplot2绘制饼图 | 69-ggplot2绘制条形图 |
70-ggplot2绘制go条目图 | 71-ggplot2绘制箱线图和小提琴图 | 72-ggplot2绘制地图 |
73-绘制美国犯罪地图 | 74-使用positron进行R语言绘图 | 75-使用jupyter绘图 |
0-rplot.zip |
13-python数据分析
| ||
1-课程介绍 | 2-直接安装python | 3-安装miniconda3 |
4-安装和配置vscode | 5-macos系统配置python环境 | 6-如何运行python |
7-在jupyter中进行数据分析 | 8-常用markdown语法 | 9-python数据分析模块 |
10-使用pip管理python模块 | 11-使用conda管理python模块 | 12-如何加载python模块 |
13-使用python模块 | 14-python数据分析书籍推荐 | 15-使用AI辅助学习 |
16-数据类型 | 17-目录结构 | 18-读写文本文件 |
19-写入文件 | 20-数据探索 | 21-读写excel文件 |
22-数据结构 | 23-删除变量 | 24-numpy常用函数 |
25-numpy数据计算 | 26-测试计算机性能 | 27-numpy索引和切片 |
28-Series数据结构 | 29-Series索引和切片 | 30-Categorical数据类型 |
31-DataFrame数据结构 | 32-纸质版表格输入到DataFrame中 | 33-DataFrame属性和方法 |
34-DataFrame索引和切片 | 35-逻辑值索引 | 36-数据类型转换 |
37-处理缺失值 | 38-python实现vlookup | 39-添加删除行列 |
40-按行列计算 | 41-修改数据 | 42-批量修改数据 |
43-排序 | 44-筛选 | 45-随机抽样 |
46-生成扑克牌斗地主 | 47-数据合并 | 48-分类汇总 |
49-数据透视表 | 50-长宽数据转换 | 51-合并拆分列 |
52-探索who数据集 | 53-python数学计算 | 54-计算频数 |
55-独立性检验 | 56-相关性检验 | 57-正态分布t检验 |
58-非参数检验 | 59-python绘图系统 | 60-python绘图网站 |
61-matplotlib绘图语法 | 62-figure绘图对象 | 63-axes绘图对象 |
64-绘图布局 | 65-使用subplots进行绘图布局 | 66-seaborn绘图 |
67-seaborn绘图设置 | 68-seaborn选项参数 | 69-可视化关联数据 |
70-可视化关联数据 | 71-可视化分类数据 | 72-绘制热图 |
73-plotnine绘图 | 74-图形语法 | 75-保存图形 |
76-绘图布局 | 77-scale修改颜色 | 78-scale修改坐标 |
79-scale修改透明度和点线形状 | 80-stat统计变化 | 81-coordinate坐标系 |
82-layer图层 | 83-叠加图层分组小提琴图 | 84-facet分面 |
85-theme主题 | 86-自定义主题 | 87-更改图例 |
88-企鹅数据集分析 | 89-MovieLens1M数据分析案例 | 90-BabyNames数据分析案例 |
91-statsmodels统计建模 | 92-线性回归 | 93-逻辑回归预测乳腺癌 |
94-方差分析 | 95-利用DeepSeek进行数据分析 | 96-利用DeepSeek处理文件 |
97-利用DeepSeek绘图 | 98-安装和配置Trae | 99-利用Trae生成代码 |
100-利用Trae进行数据分析 | 0-pydata.zip |
14-python机器学习
| ||
1-quarto配置 | 2-quarto基本语法 | 3-quarto使用案例 |
4-shiny简介 | 5-shiny语法 | 6-shiny前端 |
7-添加前端组件 | 8-shiny前端案例 | 9-shiny后端 |
10-使用AI生成shiny | 11-传文件应用 | 12-发布shiny应用 |
13-python机器学习简介 | 14-机器学习应用 | 15-scikit-learn包 |
16-获取机器学习数据 | 17-生成模拟数据 | 18-从文件读取数据 |
19-生成多维数组 | 20-导入导出模型 | 21-抽样 |
22-划分数据 | 23-K折交叉验证 | 24-什么是特征工程? |
25-标准化和中心化 | 26-离散数据预处理 | 27-文本数据编码 |
28-图像数据编码 | 29-假阳性与假阴性 | 30-计算分类模型指标 |
31-计算分类指标网站 | 32-使用sklearn评估分类模型 | 33-多元分类模型评估 |
34-回归模型评估 | 35-聚类模型评估 | 36-模型选择 |
37-网格搜索法 | 38-随机搜索法 | 39-什么是回归模型? |
40-简单线性回归 | 41-回归模型诊断 | 42-多远回归模型 |
43-波士顿房价分析案例 | 44-分类模型算法 | 45-决策树与随机森林 |
46-利用机器学习预测乳腺癌 | 47-乳腺癌预测模型评估 | 48-交叉验证提高准确性 |
49-利用网格搜索法优化参数 | 50-聚类算法 | 51-sklearn聚类 |
52-购物篮分析 | 53-什么是大语言模型 | 54-国产大语言模型 |
55-大语言模型在生物信息中的应用 | 56-AI幻觉 | 57-本地部署1-安装GPU驱动 |
58-本地部署2-安装Ollama | 59-本地部署3-安装DeepSeek | 60-本地部署4-在CheryStudio中使用大模型 |
61-本地部署5-在服务器中部署大模型 | 62-什么是大语言模型API | 63-订阅DeepSeekAPI |
64-如何使用大模型API | 65-大模型调参 | 66-订阅和使用通义千问API |
67-在CheryStudio添加API | 68-为应用添加前端页面 | 69-什么是Agent? |
70-什么是MCP? | 71-使用coze创建AI应用 | 72-安装geminicli |
73-利用geminicli进行生物信息分析 | 74-深度学习框架 | 75-安装和使用pytorch |
76-使用开源模型 | 77-张量 | 78-深度学习训练案例 |
0-pythonML.zip |
15-python生物信息编程
| ||
1-什么是软件? | 2-python语言简介 | 3-为什么要学习python编程? |
4-选择python解释器 | 5-选择python发行版本 | 6-选择python集成开发环境 |
7-安装miniconda | 8-安装vscode | 9-vscode配置python环境 |
10-python远程开发 | 11-AI编程 | 12-positron |
13-如何运行python | 14-利用AI辅助编程 | 15-使用cursorAI编程 |
16-安装和使用TraeAI | 17-使用TraeAI编程 | 18-python语法风格 |
19-变量 | 20-点号的作用 | 21-for循环 |
22-批量生成生物信息代码 | 23-while循环 | 24-AI编程完成循环 |
25-判断 | 26-循环加判断 | 27-循环控制 |
28-通过判断筛选数据 | 29-接收用户输入 | 30-os模块处理目录 |
31-利用open读入文件 | 32-按行处理 | 33-while循环处理文件 |
34-统计基因长度 | 35-读取压缩文件 | 36-写入文件 |
37-过滤blast结果 | 38-fastq格式转换为fasta | 39-开发分析流程 |
40-添加选项参数 | 41-使用argparse添加选项参数 | 42-添加长选项参数 |
43-给程序添加选项参数 | 44-python模块简介 | 45-python模块管理工具 |
46-使用pip管理python模块 | 47-批量安装模块 | 48-使用conda管理python环境 |
49-自定义模块 | 50-字符串处理1 | 51-字符串处理2 |
52-列表 | 53-读取文件为列表 | 54-fasta文件存储为列表 |
55-列表操作1 | 56-列表操作2 | 57-字典 |
58-读取文件为字典 | 59-通过字典筛选数据 | 60-字典操作 |
61-字典操作2 | 62-提取序列 | 63-试用AI生成提取序列程序 |
64-配置Copilot | 65-使用Copilot生成代码 | 66-面向对象编程 |
67-面向对象案例 | 68-创建复杂类 | 69-类的继承 |
70-面向对象统计fasta程序 | 71-什么是正则表达式? | 72-模式匹配 |
73-添加匹配flag | 74-匹配拆分序列 | 75-匹配替换 |
76-统计fasta程序 | 77-统计fastq文件 | 78-翻译氨基酸程序 |
79-AI生成翻译氨基酸程序 | 80-biopython | 81-使用biopython编写程序 |
0-python.zip |
