 夜雨聆风
夜雨聆风

工业海水电解在实现高催化活性和长期耐用性方面仍然受到限制,主要限制因素包括催化剂层内的结构降解和机械不稳定性。
清华大学王训教授、刘清达、肖海副教授等人展示了一种自粘高熵氧化物亚纳米线催化剂,有效克服了上述两大瓶颈。该催化剂在温和的条件下合成,将14种金属元素整合到均匀的~1.2 nm亚纳米线中,与导电衬底具有很强的内在附着力,无需外部粘合剂。它还具有非常规的活性位点,可实现高效、持久的晶格氧活化,同时在长时间运行中保持结构完整性。
在1 M KOH和1 M KOH+海水中,在10 mA cm-2下,过电位分别为129 mV和153 mV;可在1000 mA cm-2下连续工作4700 h和4400 h。集成到阴离子交换膜海水电解槽中,在1.70 V(80℃)下电流密度高达3000 mA cm-2,并在室温条件下、在2000 mA cm-2下连续运行超过3819小时。
相关工作以《Self-adhesive high-entropy oxide sub-nanowire monolithic electrocatalysts》为题在《Nature Nanotechnology》上发表论文。
值得注意的是,从Peer review文件上可以看到,该研究在投稿、审稿阶段遇到审稿人不同的意见。
其中,审稿人1认为:虽然文章提出的HEO催化剂性能非常出色,但在OER文献中,水热/溶剂热合成HEO路线已经很常见;同时,除了元素种类和自粘附以外,作者必须阐明其他的新发现。审稿人不同意考虑在这个杂志上发表该研究。
相比之下,审稿人2给出的评价非常高,他认为文章介绍了一种极具创新性的超高熵氧化物催化剂,其具有出色的自粘性,兼具卓越的活性和极佳的长期耐用性。作者们有力地证明,该催化剂即使在恶劣的海水环境中也能保持其性能,所达到的指标在该领域中也是名列前茅的。总体而言,这项工作在材料设计和实际应用方面都取得了重大进展。这项工作适合发表。

面对审稿人1提出的问题,作者回答道:有必要明确指出,本研究的核心创新之处并不在于单纯增加组成元素的数量,也不在于将自粘性作为一种孤立的材料特性来呈现。相反,本研究建立并实验证明了一种此前未被报道过的自粘性高熵氧化物亚纳米线一体式电催化剂的范式,在这种范式中,材料的稳定机制、电极的构建模式以及在实际运行条件下的催化行为在亚纳米尺度上是内在统一的,而非通过后期整合或外部结构工程来实现的。
如下图所示,该系统的新颖性并非来自独立设计元素的简单叠加。相反,它是从三个层次中产生的,这些层次在亚纳米尺度的高熵驱动框架内相互诱导、共同进化和相互加强。
在经历第一轮返修后,进入第二轮审稿,审稿人1仍然保持不同意发表的观点。他认为:
1、为什么修订版中作者身份有所更改?
2、作者是如何通过实验手段来控制并优化HEO中所有元素的比例,从而获得最佳的催化性能的。
3、在碱性介质中的电化学测量过程中,铝通常会严重溶解。那么铝的作用是什么呢?
4、由于实验测量结果未进行重复验证,因此该电化学方法的可靠性无法得到确认。

最后,作者花了较长的篇幅(前后花了62页PDF)、点对点地回应了审稿人提出的问题,使得文章得以成功接收、发表。
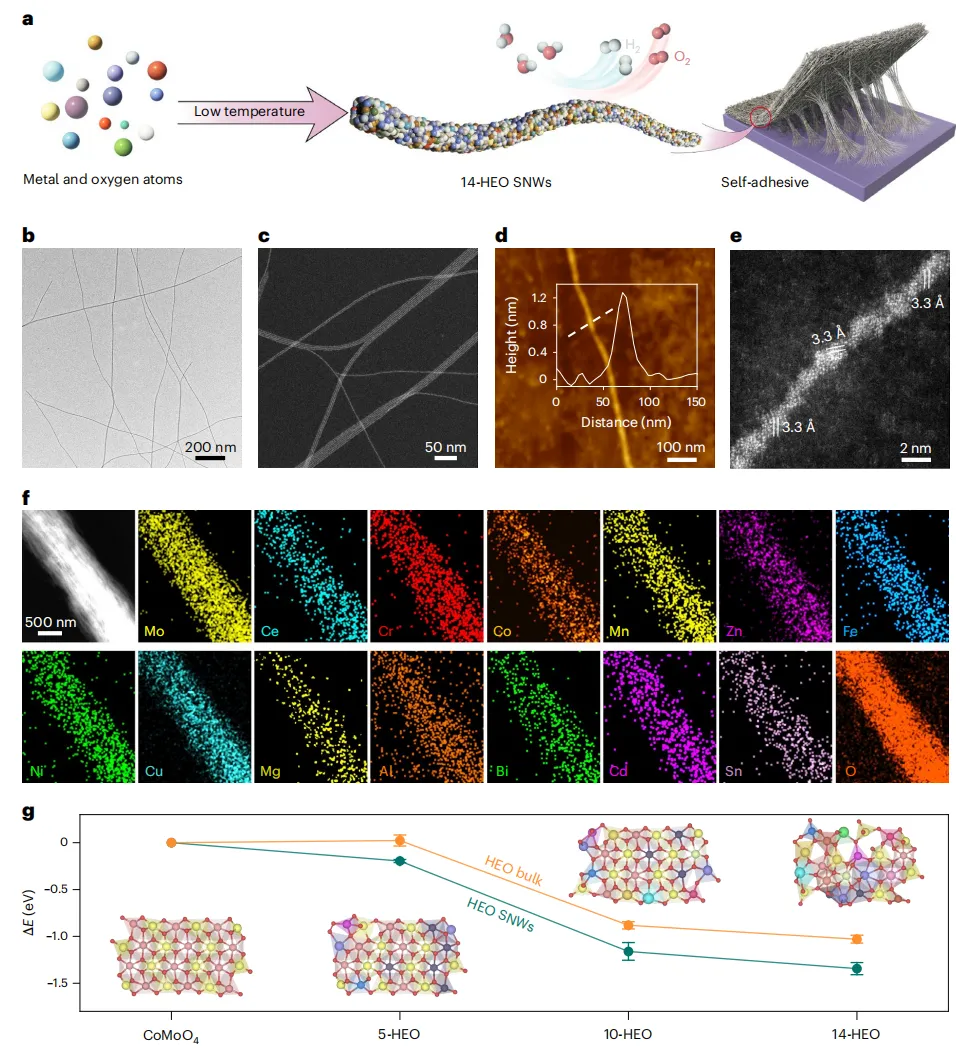
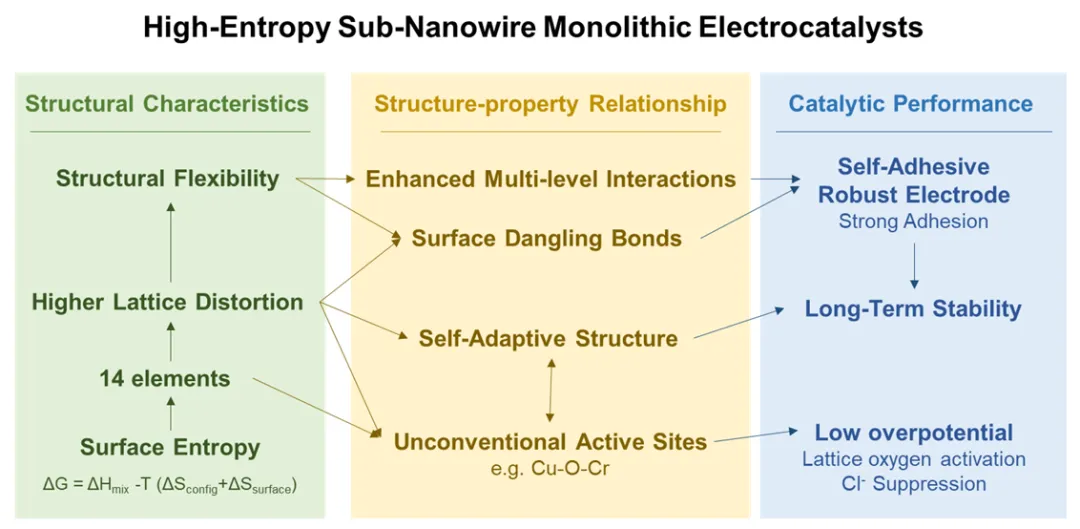
图1 14-HEO SNWs的合成及结构表征
本文报道了一种自粘高熵氧化物亚纳米线(HEO SNW)电催化剂,可在工业海水电解条件下实现高效和持久的析氧反应(OER)。该催化剂具有亚纳米、多组分结构,与导电基底具有很强的内在附着力,消除了对聚合物粘合剂的需求,提高了机械稳健性(图1a)。这种设计将结构稳定性与高催化活性结合在一起,通过稳定非常规的多金属活性位点,促进晶格氧活化,同时在恶劣的电化学条件下保持结构完整性。
如图1a所示,通过低温途径合成了14元素高熵氧化物亚纳米线(14-HEO SNWs),并表现出与基底固有的自粘附性。在温和的条件下,组成均匀的HEO SNWs的形成是由表面熵主导的热力学和受限的一维生长引起的,这有利于多种金属元素在单相中结合。采用Mo、Ce、Cr、Co、Mn、Zn、Fe、Ni、Cu、Mg、Al、Bi、Cd和Sn合成了14-HEO SNWs。值得注意的是,即使有14种组成元素,SNWs的形态和结构完整性仍然保持得很好(图1b、c)。
原子力显微镜测量(图1d)进一步证实了SNW直径约为1.2 nm,与TEM观察结果一致。X射线衍射图表明,14-HEO SNWs保持CoMoO4的单相结构,未检测到相分离。原子分辨率像差校正的HAADF-STEM图像(图1e)显示了直径约1.2 nm的超细纳米线,其面间距为3.3 Å,对应于CoMoO4的(002)面。EDS元素映射和线扫描分析(图1f)证实,所有14种金属元素在SNWs内均匀分布,未观察到可检测的聚集或相分离。
利用DFT计算研究了HEO SNWs的形成机制和元素在其中的作用。如图1g所示,HEO SNWs的热力学稳定性明显高于它们的体相。随着元素多样性的增加,稳定能逐渐降低,表明组成复杂性对SNW结构的稳定起着核心作用。这种增强的结构稳定性源于SNWs的独特性质,其低结晶度和高表面积的特点,促使金属阳离子优先占据边缘或近边缘位置,从而实现自适应的局部配位调整,协同多种键构型,增强结构的稳定性。
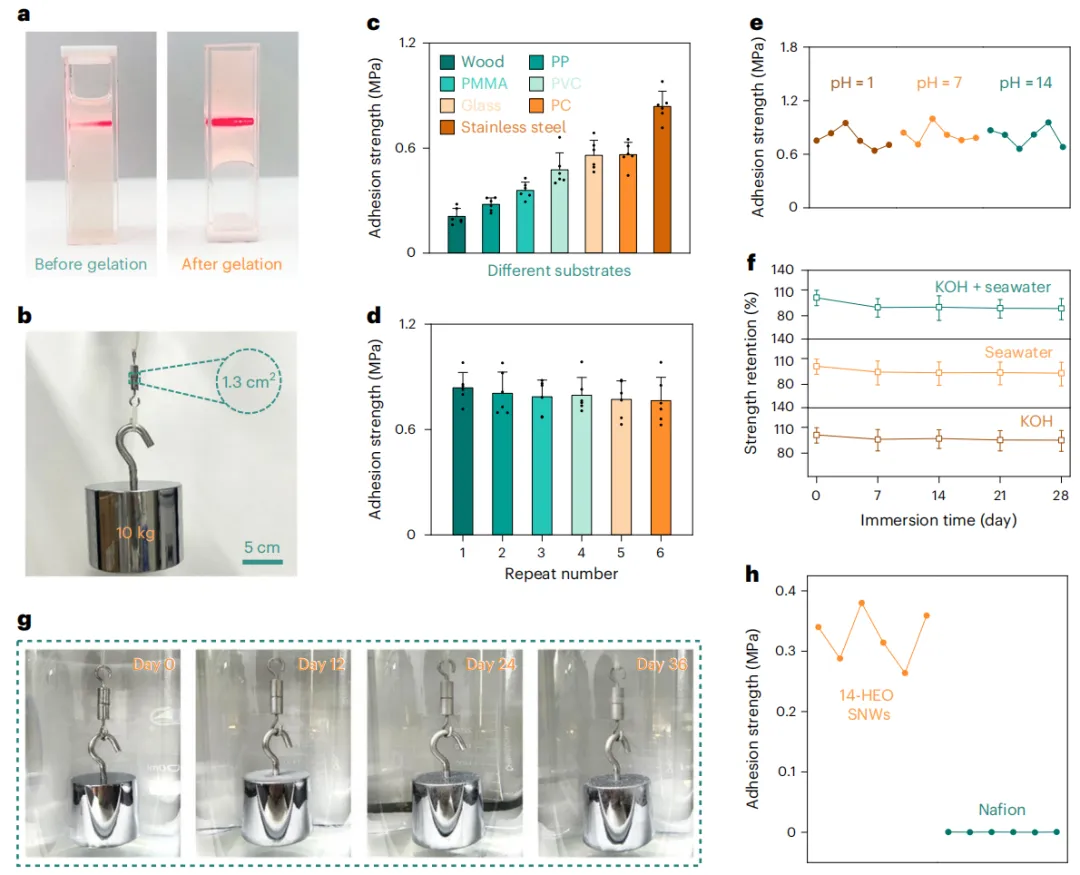
图2 14-HEO SNWs的自粘性能
由于其超细的尺寸,14-HEO SNWs表现出类似聚合物的行为。当分散在溶剂中时,它们形成一个纠缠的网络,显着增加了系统的粘度并有效地捕获了气泡(图2a)。随着时间的推移,分散体限制了溶剂的流动,形成了一种凝胶,当倒置时仍然保持完整,显示出它的高粘度。分散体和生成的凝胶都表现出明显的廷德尔效应,表明形成了一个稳定、分散良好的胶体系统(图2a)。高熵组成和SNW结构的结合导致晶格畸变和大量表面缺陷,增加了不饱和金属配位位点和M-O悬空键的密度,从而产生了与聚合物粘合剂相当的强内在附着力。在宏观测试中,14-HEO SNWs的自粘特性是显而易见的:仅由14-HEO SNWs连接的两个重物可以支撑高达10 kg的负载而不会出现滑移或断裂(图2b)。
为了评估这种粘合的普遍性,系统地评估了在一系列基底上的粘合性能(图2c)。SNWs在不同表面表现出强大的界面粘附力,在不锈钢上的强度为0.84 MPa,在玻璃和聚碳酸酯(PC)上的强度为0.56 MPa,在聚氯乙烯(PVC)上的强度为0.48 MPa,在聚甲基丙烯酸甲酯(PMMA)上的强度为0.36 MPa,在聚丙烯(PP)上的强度为0.28 MPa,在木材上的强度为0.21 MPa。值得注意的是,在重复机械应力下,粘接强度保持稳定。经过六个粘附循环后,没有观察到明显的性能损失(图2d),表明机械可靠性高。为了进一步评估电解质下的耐久性,在pH值为1、7和14的水环境中测试了粘附强度(图2e)。在所有情况下,该值都保持在0.77-0.81 MPa之间,与室温条件下的测量值几乎相同,显示出对pH诱导降解的优异抵抗能力。
通过1 M KOH、天然海水和1 M KOH+海水电解质的混合长期浸泡试验,进一步验证了其他环境下的稳定性(图2f)。7天后,不锈钢上的附着力分别保持了初始值的94.6%、93.2%和88.7%,28天后逐渐稳定,表现出优异的耐腐蚀性和环境稳定性。在KOH+海水系统中进行的宏观粘附测试证实了这种抗碱和耐盐性,其中两块不锈钢板与14-HEO SNWs粘合在一起,在超过36天的时间里持续承受1 kg的载荷,没有出现明显的脱落(图2g)。这些结果表明14-HEO SNWs在高碱性和海水环境中具有很强的粘附性,表明其在水下催化方面具有很强的应用潜力。为了测试实际性能,将14-HEO SNWs的粘附强度与商业粘合剂Nafion的粘附强度进行了比较(图2h)。14-HEO SNWs的平均粘附强度为0.324 MPa,约为Nafion的0.00033 MPa的982倍。这种显著的增强强调了它们作为无粘结剂、机械坚固的催化剂层的潜力,为传统电极在水电解中的分层问题提供了新的解决方案。
图3 14-HEO SNWs的水氧化和海水氧化性能
作者使用标准的三电极设置系统地评估OER活性。如图3a所示,催化活性随着加入金属元素数量的增加而增加。在10 mA cm-2下,14-HEO SNWs的活性最高,过电位为129 mV。为了阐明催化机理,采用原位拉曼光谱法对反应过程中的结构演变进行了监测。结果表明,在阳极极化作用下,HEO SNWs发生了电化学重构成金属羟基氧化物的过程。使用H218O的原位微分电化学质谱(DEMS)进一步证实了晶格氧的参与,检测到32O2、34O2和36O2,并且在更高的电位下增加了34O2/32O2和36O2/32O2的比率(图3b)。这些结果共同表明,OER主要通过LOM进行。
在工业电流密度下的长期耐用性对实际应用至关重要。14-HEO SNWs在1000 mA cm-2下稳定运行超过4700小时,没有明显的降解,优于大多数报道的基于LOM的催化剂(图3c、d)。该催化剂在碱性海水中表现出类似的稳定性能。电极的活性在海水中下降(图3e),主要是由于海水中存在的小颗粒、微生物和不溶性沉淀物对活性位点的侵蚀。14-HEO SNWs在306 mV过电位下达到1000 mA cm-2,远低于480 mV氯析出反应(CER)的阈值,使其成为先进的海水OER催化剂之一。金属元素多样性与催化性能之间的正相关关系在海水中保持一致。
尽管存在恶劣的腐蚀环境,该催化剂在1000 mA cm-2下保持稳定性能超过4400 h,没有明显的降解(图3f)。根据所建立的评价指标,计算出的DV为0.41 μV h-1,活性降解率(DA)为3×10-7 h-1(图3g),大大低于美国能源部(DOE)的目标,优于其他已报道的催化剂(图3h)。法拉第效率测量证实了近100%的O2选择性,没有检测到与氯相关的氧化产物(图3i)。与最先进的海水OER系统相比,14-HEO SNWs具有更长的使用寿命和活性(图3j)。这些结果清楚地表明OER是海水电解条件下的主导过程。
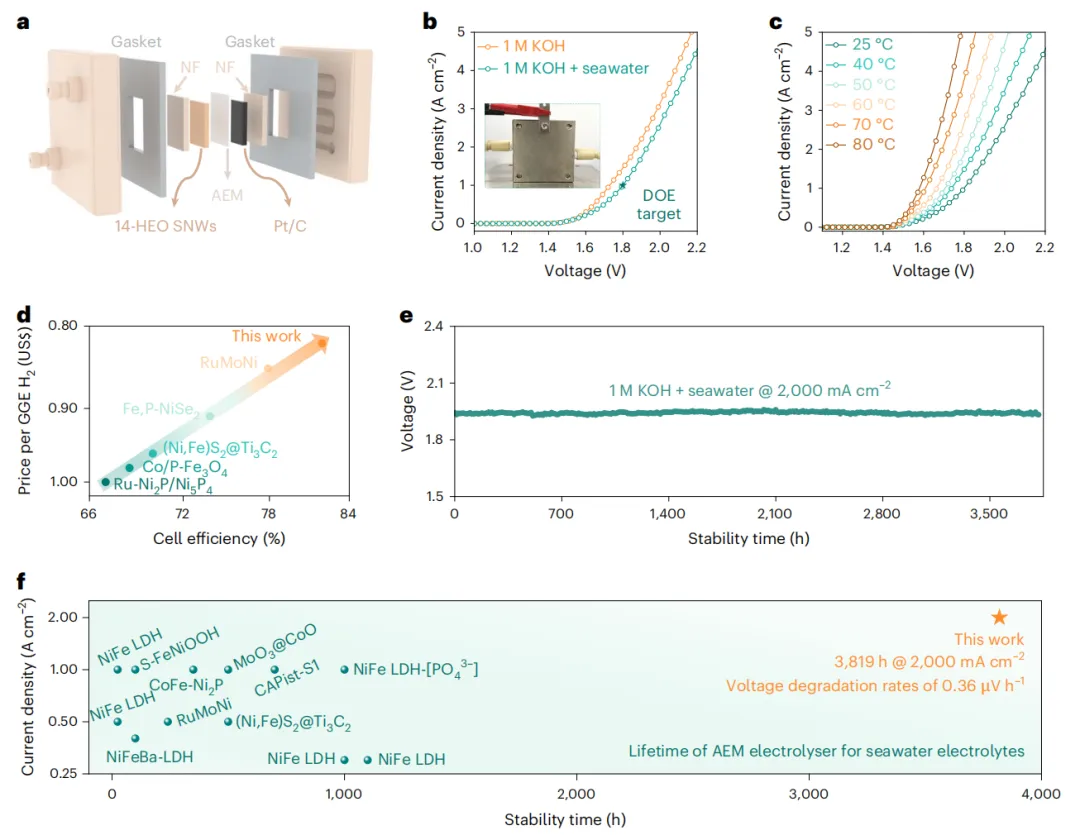
图4 基于14-HEO SNWs的AEMWE/AEMSE器件性能
为了评估实际适用性,以14-HEO SNWs为阳极组装了一个阴离子交换膜水/海水电解槽(AEMWE/AEMSE)(图4a)。该器件在1 M KOH和1 M KOH+海水下分别以1.73 V和1.80 V的电压在室温下获得1000 mA cm-2,满足美国能源部对AEMWE系统的目标(图4b),显示出工业级的性能。升高温度进一步改善活性;AEMSE在1.58 V和1.70 V时分别达到1000 mA cm-2和3000 mA cm-2,这是迄今为止报道的性能最好的系统之一(图4c)。
该系统还实现了81.9%的能源效率,制氢成本低至每加仑汽油当量(GGE)0.82美元,大大低于美国能源部2026年2.00美元的目标(图4d)。AEMSE具有优异的耐久性,在室温条件下,在2000 mA cm-2下保持稳定运行超过3819小时(图4e),最小DV为0.36 μV h-1。值得注意的是,在高电流密度下,这种水平的长期稳定性在AEMSE系统中名列前茅(图4f)。这些结果表明,自粘14-HEO SNW催化剂具有高性能、长期耐用性和工业规模制氢的经济可行性。
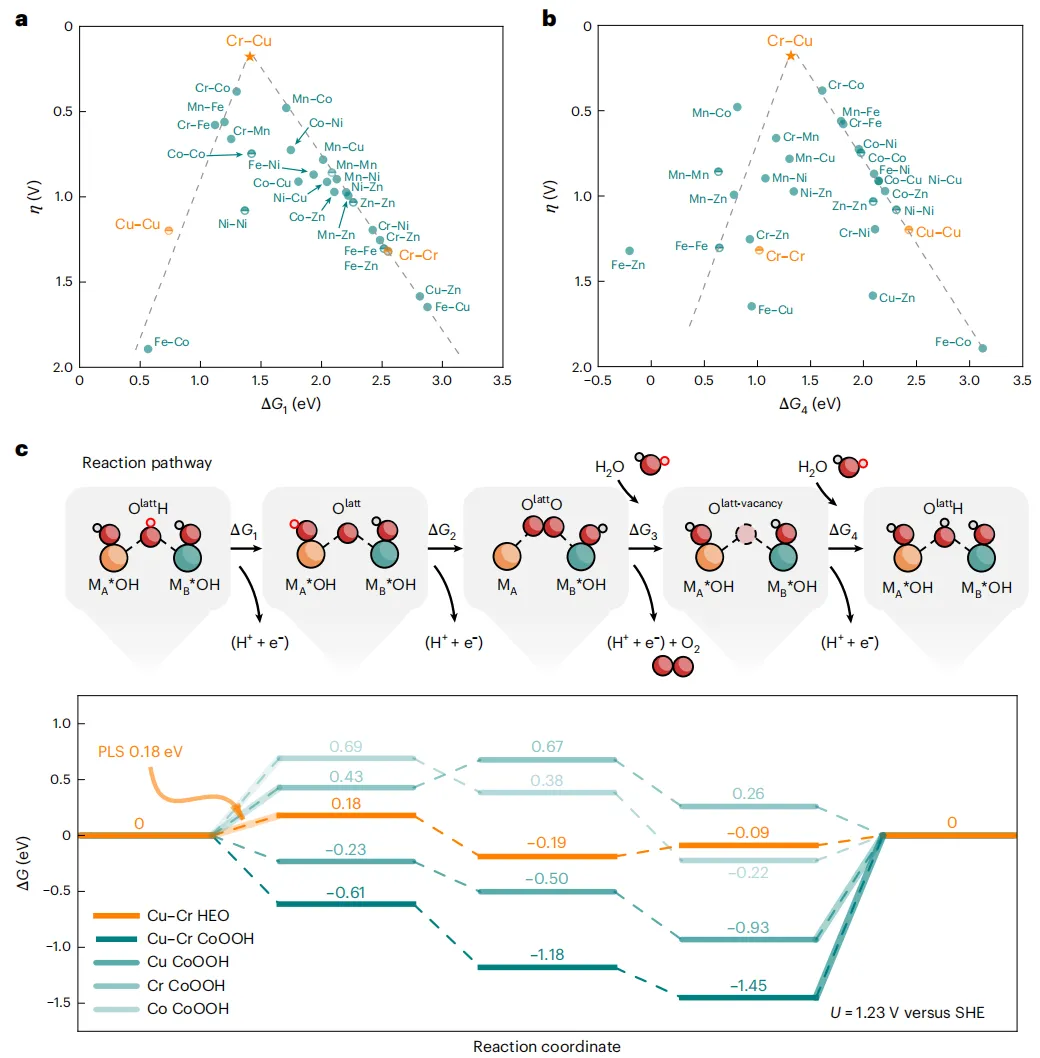
图5 DFT计算
之前的研究表明,在一维亚纳米羟基氧化物结构中,边缘位置的双金属中心MA-O-MB是关键的活性位点,促进了通过LOM途径的OER。在这项工作中,HEO SNWs采用了相同的途径,并构建了火山图,以确定活性中心的最佳元素组成。由于LOM涉及晶格氧的直接参与,其固有活性与金属物种的成键性质有关。因此,考虑了两个重要的电化学步骤(图5a、b),这两个步骤分别说明了晶格氧的内在活化能力和基于稳定性的恢复能力。在通过完整的OER路径评估了多种可能的双金属中心组合后,证明了Cr-O-Cu在稳定和激活晶格氧之间达到了最佳平衡。这种增强源于不同金属阳离子和晶格氧之间的键合强度的差异。随着原子序数沿3d过渡金属系列的增加,M-O键特征逐渐从离子键转变为共价键,有效地描述了激活和稳定晶格氧的能力。
图5c给出了OER的能量分布,其中Cu-O-Cr的电位限制步骤(PLS)是晶格位点OlattH (0.18 eV)的去质子化,对应于计算的过电位(η)为180 mV。除了提供这些活性位点外,HEO SNWs还可以作为基底发挥额外的作用。为了理解这一点,考虑了以CoOOH取代HEO作为基底的模型催化剂(图5c),在这些情况下,CoOOH上的Cu-O-Cr位点和Cu或cr掺杂的CoOOH都没有表现出HEO SNWs中Cu-O-Cr活性位点的优越催化活性。有趣的是,HEO和CoOOH上的Cu-O-Cr活性位点显示出OER活性的显著差异,这强调了HEO基底的关键作用。
Self-adhesive high-entropy oxide sub-nanowire monolithic electrocatalysts,Nature Nanotechnology,2026.
https://www.nature.com/articles/s41565-026-02175-4


