 夜雨聆风
夜雨聆风2025年6月,FDA全机构上线了内部AI助手Elsa——它能识别高风险检查目标、汇总不良事件趋势、辅助临床方案审查。本文拆解Elsa和FDA其他四个AI工具的真实架构,然后回答一个实操问题:中国药企能不能参照这套逻辑,给自己部署一套内部质量审计AI?
开篇:这件事比它看起来更值得认真对待
2026年2月27日,特朗普签署行政令,要求所有联邦机构停止使用Anthropic的Claude,理由是国防部与Anthropic在"AI用于自主武器"问题上爆发了严重争议,国防部长将Anthropic列为"国家安全供应链风险"。结果是:FDA刚上线8个月的核心AI工具Elsa,被迫开始迁移至Google Gemini。(来源:Clinical Leader,2026年3月12日;白宫行政令,2026年2月27日)
这件事之所以值得药企管理层认真对待,不是因为AI底座换了谁,而是因为它说明了一件事:Elsa已经深度嵌入FDA的日常运营,嵌入到"换底座"这个技术操作需要专程向行业发出风险警告的程度。这不是一个内部效率工具,这是一个已经影响检查优先级排序的监管决策辅助系统。
对中国药企来说,这个信号的含义很具体:FDA用AI找高风险检查目标的能力,正在快速强化。传统上,飞行检查的触发逻辑主要依赖人工风险判断和周期性轮检计划;现在,Elsa能在数小时内跨数据库识别你的合规异常信号,而这个周期以前是数月。
这篇文章做两件事:第一,逐个拆解FDA目前在用的五个AI工具,评估哪些逻辑可以被药企内部借用;第二,基于Elsa的架构原理,给出一套可落地的企业质量审计AI部署框架。
一、FDA目前在用的AI工具:五个,逐个拆解
Elsa:全机构大语言模型助手(2025年6月上线)
Elsa(Electronic/Enterprise Language Support Assistant)由咨询公司Deloitte开发,从CDER内部试点工具CDER-GPT演进而来,于2025年6月2日全机构上线,提前完成且低于预算。(来源:FDA官方新闻稿,2025年6月2日)Deloitte在此项目上分两期获得合同:第一期$1380万用于构建FDA内部文件数据库,第二期$1470万用于全机构扩展。(来源:STAT News/BioSpace交叉核实)
Elsa的架构是定制化RAG(检索增强生成)系统,部署在AWS GovCloud的FedRAMP High认证环境中。这不是一个通用聊天工具——它整合了FDA内部的文件库、嵌入模型、向量数据库和专为监管场景调优的提示词模板,整套系统原本针对Anthropic Claude的行为特性专门设计。(来源:Clinical Leader,2026年3月)
FDA公开披露的Elsa功能包括:汇总不良事件报告趋势、比较产品标签版本、辅助临床方案审查、识别高风险检查目标、以及生成内部数据库查询代码。其中"识别高风险检查目标"是对药企影响最直接的功能——Elsa通过分析FDA内部的不良事件报告、Form 483观察项、历史检查结果和合规数据异常,对检查目标进行优先级排序。(来源:FDA官方声明;Reed Smith律所合规分析,2025年12月)
数据隔离机制需要单独说明:Elsa不接入企业提交给FDA的NDA/BLA/ANDA数据,不在行业提交文件上训练。这个设计一方面是合规要求,另一方面也决定了Elsa的判断依据完全来自FDA内部记录——也就是说,你的历史检查记录、你提交的不良事件报告、你的Form 483历史,都在它的分析范围里。
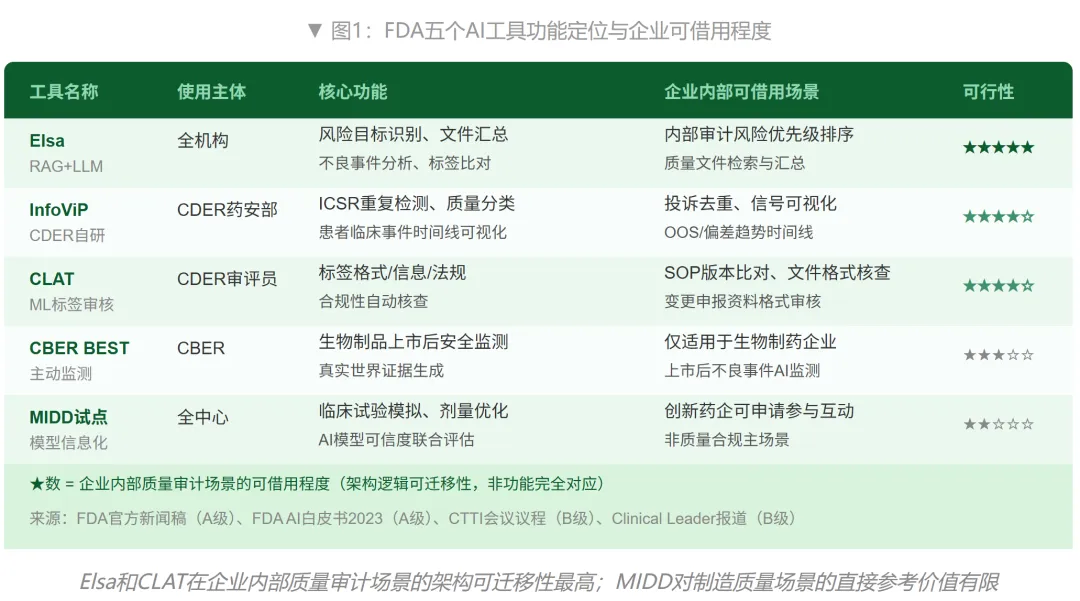
InfoViP:CDER自研的个案安全报告分析平台(文献记录最完整的一个)
InfoViP(Information Visualization Platform)是CDER药物安全监测办公室自主研发的AI工具,专门处理FAERS(FDA不良事件报告系统)中的个案安全报告(ICSRs)。三类核心任务:检测重复提交的ICSRs避免重复计数、按信息完整度对报告质量分类、以及可视化患者临床事件时间线辅助信号判断。项目负责人Oanh Dang在多篇同行评审期刊上发表了相关研究,这是FDA五个AI工具中文献记录最系统的一个。(来源:FDA官网CDER EDSTP页面;CTTI 2025年研讨会议程)对企业的参考价值在于:投诉去重、质量信号时间线可视化,这两个逻辑可以直接迁移到企业投诉处理和偏差趋势分析中。
CLAT:标签自动化审核(最接近"文件合规AI"的一个)
CLAT(Computerized Labelling Assessment Tool)用机器学习自动审核药品标签的格式错误、信息缺失和法规不一致。(来源:FDA 2023年AI白皮书)这个工具的逻辑对企业内部文件合规核查高度可迁移——用同样的方法做SOP新旧版本差异分析、变更申报资料格式核查、以及批记录与规程要求的一致性比对,是企业质量合规AI中性价比最高的场景之一。
CBER BEST和MIDD:相对专门化,企业直接借用空间有限
CBER BEST系统针对生物制品的上市后安全主动监测,通过分析电子健康档案预测不良事件。(来源:FDA官网CBER BEST IM页面)这对生物制药企业的药物警戒AI有参考价值,但对一般制剂制造企业的质量审计场景直接可用性有限。MIDD试点项目主要面向研发阶段的AI/ML应用,与质量合规审计场景的交集较小,但对正在推进FDA申报的创新药企,MIDD是目前唯一可以和FDA就AI模型可信度评估标准进行正式沟通的渠道。
二、Elsa对企业质量审计的真实影响:不只是"被更严格地查"
理解Elsa对企业的影响,需要先理解它实际上在做什么。在Elsa之前,FDA的检查优先级排序本质上是一个人工+数据驱动的混合流程:审评员需要跨越FAERS数据库、历史检查记录、Form 483档案多个系统手工提取信号,这个过程可能需要数天。现在,同样的过程可能在数小时内完成,并且能识别以前因数据量太大而被忽略的"轻微但系统性的漂移"。(来源:FDA Group Insider,2025年6月)
对中国出口FDA监管市场的药企,影响是具体的:一次客户投诉中的产品污染描述,一批不良事件报告中某种杂质的反复出现,一家工厂跨批次的OOS模式,这些信号现在会在更短的时间内被关联和评分。以前靠"检查时间窗口"掩盖的质量漂移,会越来越早进入FDA的优先队列。(来源:Reed Smith合规分析,2025年12月)
这里有一个从FDA Elsa架构能读出来的、但很少被直接说出来的结论:Elsa对企业的本质压力,不是让FDA查得更多,而是让FDA查得更准。以前可能在轮检计划轮到才会触发的检查,现在因为合规数据异常信号会被提前拉进优先队列。这个变化对合规历史干净的企业几乎没有影响,但对有未解决的历史Form 483观察项、或存在系统性CAPA未关闭的企业,是一个明显的压力提升。
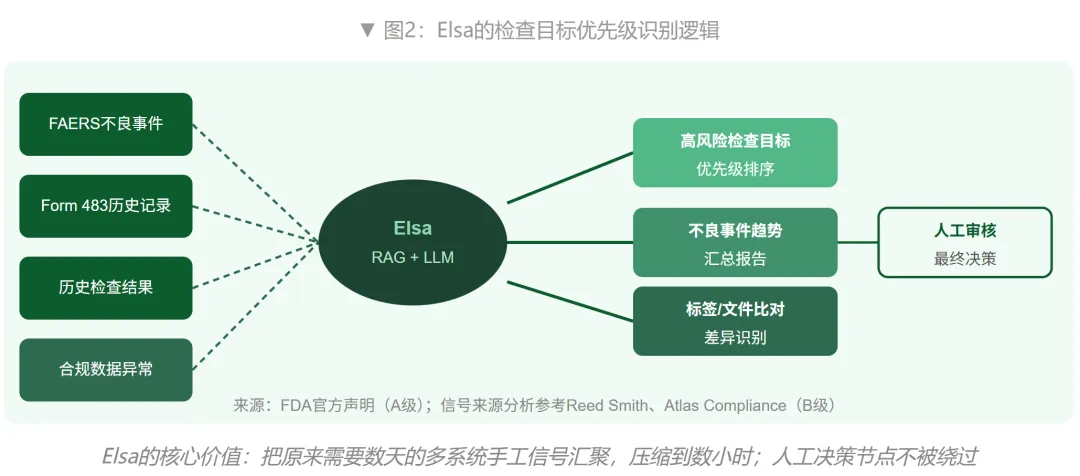
三、给企业部署一套"内部Elsa":架构逻辑与四个核心模块
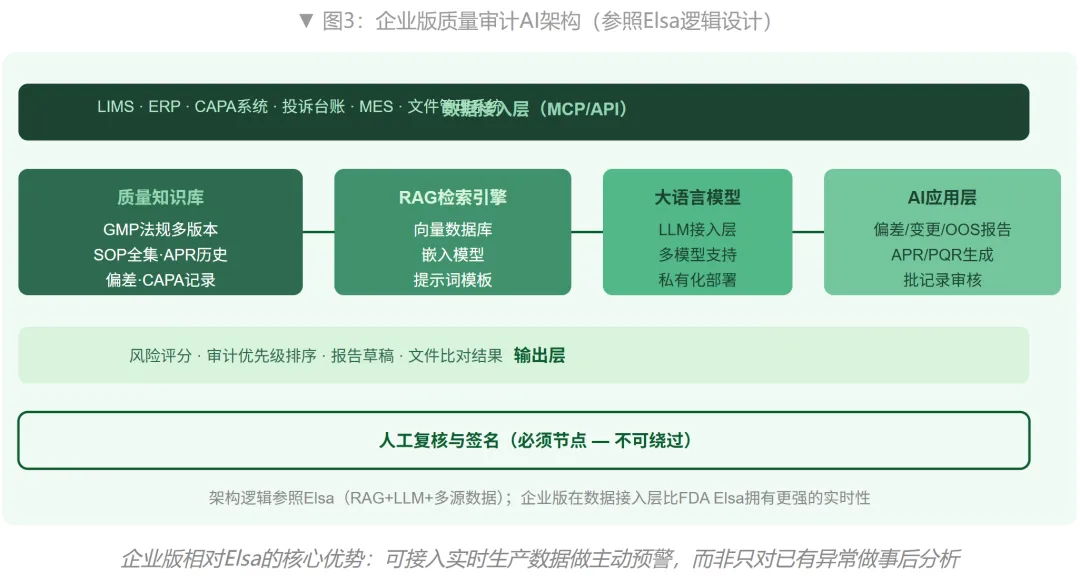
理解了Elsa的架构之后,一个很自然的问题是:同样的逻辑,能不能在企业内部复现?答案是肯定的,而且企业版在某些维度上更有优势——企业能接入Elsa接不到的数据(ERP、MES、LIMS实时数据流),能做FDA做不到的事(对未发生的质量风险进行主动预警,而不只是识别已有信号)。
企业版"质量审计AI"的核心逻辑,和Elsa高度同构:都是RAG架构,都是多源数据汇聚,都是输出风险评分和文件汇总,都保留人工复核节点。差别在于:FDA的数据源是监管侧的不良事件和检查记录,企业的数据源是内部的质量体系运营数据。
以下是企业质量审计AI的四个核心模块,每个模块都有直接对应的Elsa能力作为参照。
模块一:质量知识库(对应Elsa的FDA内部文件库)
Elsa的知识库涵盖FDA内部协议、标签文件和检查报告,训练数据约12亿token。企业版的知识库应覆盖:现行GMP要求(NMPA/FDA/EMA多版本)、企业内部SOP全集、历史偏差和CAPA记录、历史批记录关键参数、产品质量年度回顾(APR/PQR)历史报告。这个知识库是整个系统的基础——没有结构化的知识库,RAG系统输出的内容就只能是通用建议,而不是针对具体产品和具体工艺的合规判断。
模块二:多系统实时数据接入(对应Elsa的FAERS+Form 483数据汇聚)
这是企业版相对FDA Elsa的关键优势:可以通过API或MCP(Model Context Protocol)连接LIMS(检验数据实时流)、ERP(批次生产数据)、CAPA系统(偏差和整改记录)、投诉管理系统。Elsa每次分析需要人工触发,企业版可以做到持续监控——当某个批次的含量检验数据出现连续下移趋势时,系统主动推送警报,而不是等到下一次定期审计才发现。
模块三:审计风险评分与智能排序(对应Elsa的检查目标优先级识别)
这是价值最大的模块,也是从Elsa架构能学到最多的部分。企业版的逻辑是:基于历史偏差频率、CAPA关闭率、批次不合格趋势、工艺参数稳定性指标,对产品线、部门、供应商进行风险评分,输出内部审计的优先级排序。高风险品种/部门优先安排内审,而不是只按照轮检计划走。这个模块能把"内审效率低、问题发现滞后"这个合规盲区系统性压缩。
模块四:审计文件自动生成与合规核查(对应Elsa的文件汇总和CLAT的标签核查)
这是最容易快速看到价值的模块:输入审计范围,AI自动从知识库中提取相关SOP、历史偏差记录、CAPA状态,生成审计准备清单和初步问题清单;审计结束后,AI根据审计员录入的观察项,自动生成符合NMPA或FDA格式要求的审计报告草稿;文件版本比对功能可以自动识别SOP新旧版本的差异及其合规影响。
四、从现有AI平台出发的落地路径:不需要从零开始
对于已经拥有医药AI平台基础设施的企业来说,部署上述四个模块不需要从零建设。以具备智能体编排、RAG知识库、MCP工具调用、Skill能力建设和多模型接入能力的AI平台为基础,企业质量审计AI的部署路径可以分三个阶段推进:
第一阶段(1—2个月):知识库建设与单点功能上线。优先把质量合规知识库搭起来——现行GMP法规、企业SOP全集、历史APR报告——这是整个系统的基础。同时,先上线单点高价值功能:偏差报告初稿生成、变更报告辅助、OOS/OOT调查引导。这些功能有现成的提示词框架,可以快速在平台上配置智能体完成。这一阶段的价值验证不依赖系统集成,可以在文档导入+对话交互模式下先跑通。
第二阶段(2—4个月):数据接入与风险评分模块建设。通过MCP或API将LIMS检验数据、CAPA系统、投诉台账接入AI平台。在数据接入完成后,开始建设风险评分模型:基于历史偏差频率、OOS发生率、CAPA超期关闭率等指标,构建产品线和部门级的风险仪表盘。这个阶段需要IT和质量团队联合参与,关键挑战是数据标准化——如果现有系统导出的数据格式参差不齐,需要先做数据治理。
第三阶段(4—6个月):完整审计AI闭环。在前两阶段基础上,补齐审计计划智能生成、现场审计问题发现辅助、以及审计报告自动生成功能。配合批记录自动化智能审核(EBR审核)和检验记录与报告书比对,形成从审计计划→现场执行→报告生成的完整闭环。这个阶段的关键是流程标准化和人机协同规范——AI输出必须有清晰的人工复核节点,且全程留有审计追踪。
我们跑通的实践:懂规矩的“AI 辅助质量审计平台”
作为专注于医药行业的AI 原生平台,打造了以LLM、Agent、RAG、Skill、MCP、Harness 技术为核心的企业级AI 平台,结合医药行业深度领域知识,构建了覆盖研发、生产、质量全链路的AI 智能体产品矩阵。
在帮两家大厂做完方案梳理后,我们没有画那种“大而全的企业大脑”,而是选择把我们自研的AI辅助质量审计平台部署过去。这个平台的底层不是大模型的聊天框,而是根据药企质量、审计和日常合规的真实流向,拆成了四个协同的专业模块。它们不代替人工做最终决策,但负责将审计全链条的文档核对与信号分析前置完成。
药企“AI 辅助质量审计平台”架构全景

Elsa被迫从Claude迁移到Gemini这件事,从技术角度来说是一个非常麻烦的工程问题——整个RAG管道(嵌入模型、向量数据库、检索逻辑、提示词模板)都是针对Claude的特性专门调优的,换底座等于需要重新验证整个系统。(来源:Clinical Leader,2026年3月)这个麻烦不是因为FDA工程师做了错误的技术选择,而是因为他们做了一个极度依赖单一外部模型提供商的架构决策。
对企业来说,这件事的教训是:在部署企业质量AI系统时,模型层的解耦设计不是可选项,是必须项。支持多模型接入(本地私有化模型、国产大模型、商业模型混用)、底层架构不锁定在任何单一供应商,这个能力在设计阶段就需要考虑进去,而不是等到需要迁移时才意识到它的重要性。
可迁移启示与我的判断
启示一:从"文件生成"开始,不从"风险评分"开始。多数企业的正确起点是AI辅助质量文件生成(偏差报告、变更报告、审计报告草稿),而不是直接上风险评分模型。原因是:文件生成的价值验证周期短(2—4周可以看到结果),不依赖复杂的系统集成,对现有工作流程的干扰最小。适用条件:所有计划引入质量AI的企业,无论规模大小。
启示二:数据治理是瓶颈,不是工具选型。Elsa能发挥价值,是因为FDA内部有高质量的结构化数据——FAERS数据库、Form 483观察项数据库、检查结果数据库,这些数据是几十年积累的规范化记录。企业版质量审计AI要发挥Elsa同等的风险识别能力,前提是LIMS、CAPA、批记录等系统里的数据是结构化、可机读的。数据基础不到位,买再好的AI工具也只能做单点文件生成,做不到跨系统的风险信号汇聚。适用条件:计划在6个月以上周期内部署完整审计AI的中大型药企。
启示三:人工复核节点不可绕过,但审核内容应重新定义。Elsa的设计明确保留人工决策节点,FDA没有让AI直接决定检查目标。这不只是合规要求,也是避免"AI幻觉进入正式监管决策"的基本安全机制。企业版应该做同样的事——AI生成的风险评分和报告草稿,必须由有资质QA人员审核签名。但"审核"的内容应该重新定义:从审核人工写作的每一句话,变成审核AI输出的逻辑是否合理、结论是否有数据支撑。这是技能提升,不是工作量叠加。适用条件:所有质量体系中引入AI辅助工具的场景。
💡 判断
FDA部署Elsa这件事,对中国药企的真实影响不是"会被更多地检查",而是"会被更准确地识别高风险信号"。最好的应对不是去读懂Elsa的算法,而是在它找到你之前,先用同样的逻辑找到自己的问题——这正是企业版质量审计AI的核心价值。参照Elsa的架构设计内部系统,而不是从零发明,这条路是对的。
数据来源说明:Elsa相关信息来源于FDA官方新闻稿(2025年6月2日)、白宫行政令(2026年2月27日)、Clinical Leader深度分析(2026年3月)、Reed Smith律所合规分析(2025年12月)、Atlas Compliance、IntuitionLabs研究报告。InfoViP来源:FDA官网CDER EDSTP页面、CTTI会议议程。CLAT来源:FDA 2023年AI白皮书。CBER BEST来源:FDA官网CBER BEST IM页面。所有来源可通过各机构官方网站独立核实。
