 夜雨聆风
夜雨聆风一、 AI给2000+分子做了一次"体检",3个维度筛出了最优解
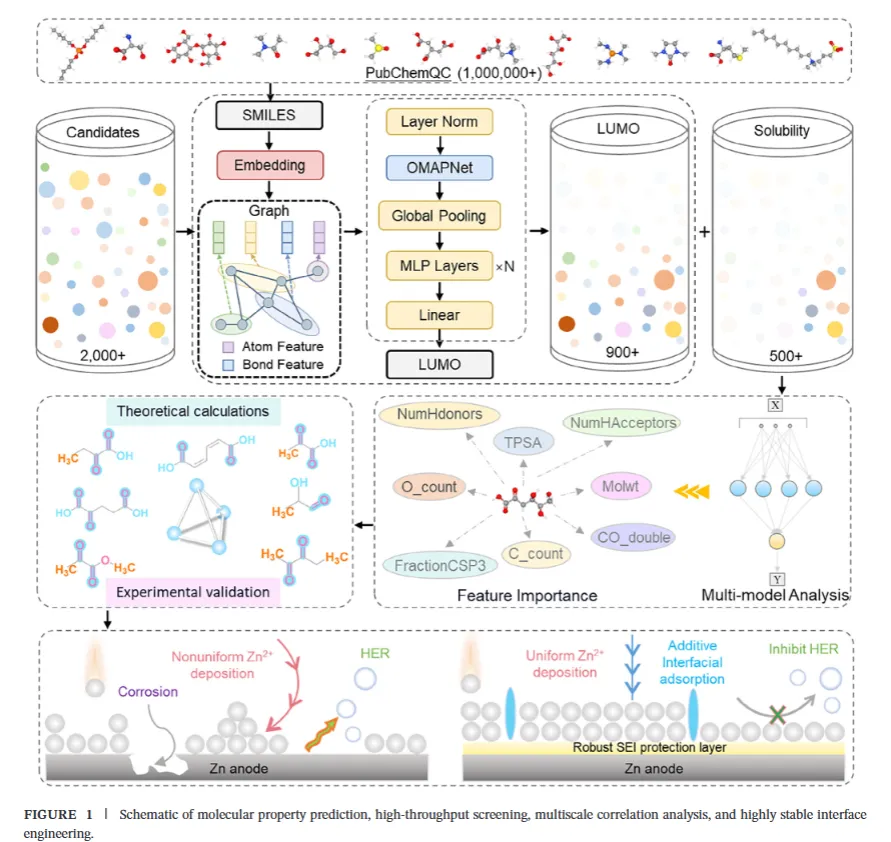
传统的添加剂筛选,基本靠"试"。一个团队合成一个分子,测试,失败,再来一个……劳动密集、资源密集、效率极低,而且选出来的分子往往只是"还行",远不是最优。
上海大学程宏伟团队(通讯作者 hwcheng@shu.edu.cn)这回换了个思路:不是盲目试错,而是先问清楚——什么样的分子,天然就适合去保护锌负极?
他们找到了一个物理量:LUMO能级。
所谓LUMO(最低未占据分子轨道)能级,你可以简单理解为:一个分子有多"渴求"电子。 LUMO越低,分子越容易接受电子,越容易在电化学环境中被还原——换句话说,越容易在锌负极表面"率先分解",形成保护层。
关键阈值是 -0.968 eV。 这是水分子(H₂O)的LUMO能级。研究者的逻辑很清晰:只要一个分子的LUMO低于这个值,它就会比水分子更先一步接受电子、在锌负极表面还原,形成一层保护膜——相当于抢在水分解之前,给负极穿上一层"皮肤"。
但光LUMO低还不够。分子还得能溶于水(logP ≤ 0),否则加进去直接沉底,发挥不了作用。
于是,研究者搭建了一套三层协同框架:
① 物理描述符:用LUMO能级和logP(溶解性)作为双筛标准,从源头确保分子"还原优先 + 水溶性合格"。
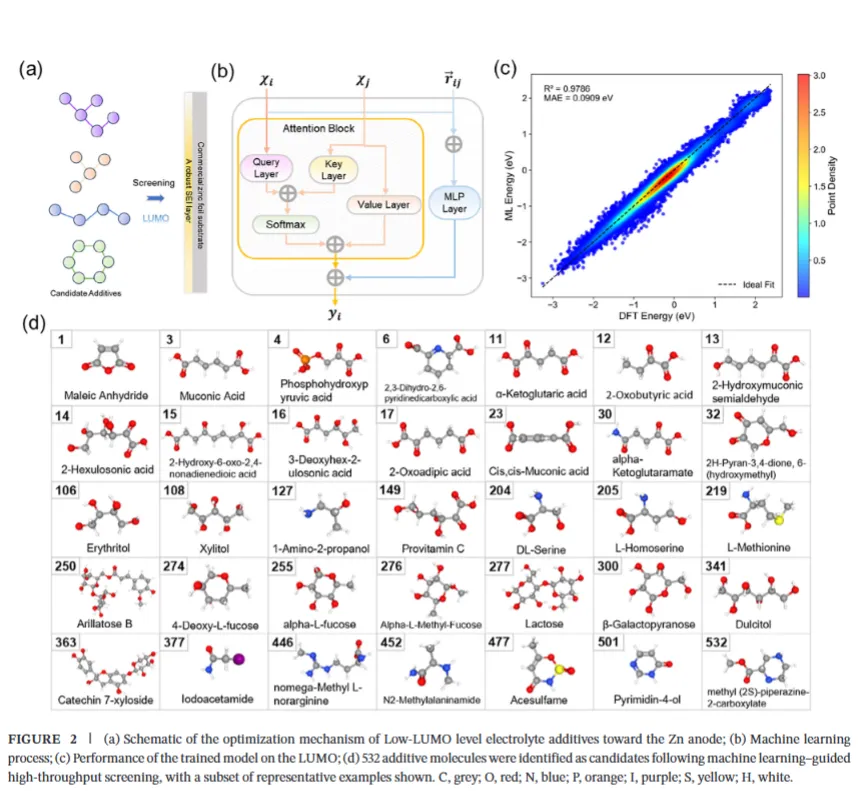
② 图神经网络(OMAPNet):构建了一个专门预测分子LUMO能级的高精度注意力图神经网络,在100万分子数据库上训练,测试集 R² = 0.9786——这个精度,相当高了。然后用它对2000+候选分子批量预测LUMO值。
③ SHAP可解释性分析:让模型"开口说话"——不是给个黑箱预测完事,而是用SHAP值量化每个分子特征对LUMO的贡献,找出"到底什么结构特征让LUMO变低"。
三层筛完,从2000+分子中锁定了532个候选,最终选定综合最优的分子:α-酮戊二酸(Ket)。
二、 最让人意外的结构发现:羰基"锁电子",效果出乎意料
Ket被选中的原因很直接:LUMO够低(-2.583 eV,远低于水的-0.968 eV),logP也符合水溶性要求。
但真正有意思的,是SHAP分析揭开的结构秘密。
SHAP特征重要性排名前六:sp³碳分数(FractionCSP3)、碳氧双键数(CO_double)、杂原子数、氢键供体数、氢键受体数、氢原子数。CO_double(碳氧双键数)排名第二——羰基(C=O)的数量,是决定LUMO能级的关键变量。
DFT计算更是直接揭示了背后的电子结构机制:
在Ket这样的α-酮羧酸分子里,两个相邻的羰基C=O紧紧挨着。这种"邻位二羰基"结构,让羰基的电子被牢牢锁在键上,形成了极其局域化的LUMO轨道——电子云高度集中,分子"吸电子"能力极强,LUMO因此降到很低。
更有意思的反常识发现: 在药物化学里,羰基有时被视为"不稳定的结构元素"。但在这项研究里,恰恰是羰基的电子局域化,让Ket成为了最理想的界面保护剂——"不稳定性"在这里变成了反应活性,让分子精准地在界面发生受控还原,形成SEI保护层。
研究者还对比了多个分子:Cis(-1.658 eV)、Ket(-2.583 eV)、Oxo(-2.430 eV)、Met(-3.002 eV)、Pyr(-2.532 eV)、Mep(-1.587 eV)、Pent(-2.453 eV)。无例外,它们的LUMO全部远低于水,而含羧酸基团(-COOH)但无邻位羰基的Mep,LUMO反而偏高(-1.587 eV)——进一步印证了:羰基的空间紧邻排布,是LUMO降低的核心结构密码。
三、 界面"皮肤"是怎么炼成的:羰基锚锌,梯度SEI来护驾
Ket是怎么保护锌负极的?整张图景逐渐清晰。
第一步:优先吸附。 Ket分子带着"饿极了"的羰基氧,吸附在Zn(002)晶面表面。DFT计算显示,Ket在Zn表面的吸附能(-2.09 eV)显著低于H₂O(-0.19 eV)——Ket比水更"黏"锌,这意味着在界面竞争里,Ket已经赢在起跑线。
第二步:界面还原,形成SEI。 随着电化学循环,Ket分子在锌负极表面发生还原分解。但这个分解不是乱七八糟的——羰基的定向吸附,让还原产物有序排列,形成一层结构精密的有机-无机梯度杂化SEI:
• 外层(有机):Ket的有机还原产物,主要作用是防水——把水分子和质子挡在外面,从源头抑制氢气析出。 • 内层(无机):ZnO + ZnS富集的纳米颗粒层——ZnS来自硫酸锌电解液与有机还原产物中硫元素的反应。这层无机层离子电导率高,专门负责让Zn²⁺快速"过关"。
这层"皮肤"有多重要?用一句话概括:它同时做到了"挡住水"和"放行离子"——这是理想SEI膜的两大核心功能。
没有这层膜:锌离子通量分布不均,枝晶疯狂生长,副反应持续消耗活性锌。
有了这层膜:界面电场重新分配,Zn²⁺在SEI内均匀分布,在Zn表面均匀成核、均匀沉积,枝晶和"死锌"问题同时被压制。
这层膜带来的连锁反应,远不止"减少枝晶"这么简单:
氢气析出(HER)被显著抑制——这意味着电池运行更安全,循环过程中不再"胀气"。
腐蚀电流从0.4639降到0.09344 mA/cm⁻²(降低80%)——锌负极的"皮肤"变厚实了,腐蚀被压住。
Zn²⁺迁移数从0.2126提升到0.5894(接近3倍)——界面离子传导效率大幅提升,大电流充放电不再卡壳。
脱溶剂活化能从31.96降到24.16 kJ/mol(降低24.4%)——Zn²⁺脱掉水分子外壳更容易了,动力学阻力减小。
数据说话:187天不间断 + 290天静置复活
对称电池(Zn||Zn)在5 mA·cm⁻² / 1.0 mAh·cm⁻²条件下:稳定循环超过4550小时,折合约187天,电压曲线全程平稳,无短路迹象。
更震撼的是静置寿命测试:静置超过7000小时(约290天)后,电池依然能恢复正常工作状态——电压滞后极低,可逆性极高。这说明什么?Ket形成的SEI"皮肤"不只是在动态充放电中有效,在长时间静置状态下,也能稳稳地保护锌负极不被电解液侵蚀。
Zn||Cu电池:3500周循环,平均库仑效率99.93%——每次充放电,能量利用率接近完美,容量损失微乎其微。
极端电流测试:30 mA·cm⁻²电流密度下稳定运行300小时以上——这个数字,比绝大多数已报道的锌负极改性方案高出一个数量级。20 mA·cm⁻²极电流密度下,同样稳定运行300小时以上。
85.5% DOD超薄锌负极:350小时稳定循环——说明即使锌源有限,Ket策略依然有效,这对实际储能应用意义重大。
全电池(Zn||NVO)表现:
• 1.0 A/g:1000次循环后容量保留78.2%,对照组仅41.98%; • 高载量10 mg·cm⁻²:600次循环后保持81.6%初始容量; • 自放电:静置24h容量保留88.9%(对照组65.7%),48h保留80.7%(对照组58.2%)。
四、 方法论破局:AI+物理描述符,正在重写电池材料设计规则
回顾整个研究,最值得关注的不只是数据本身,而是一套从分子轨道到界面化学的精准设计范式。
传统的"试错法"在这里被彻底颠覆:研究者不是盲目合成候选分子再去测性能,而是从第一性原理出发,找到了LUMO能级这个连接分子结构与界面电化学行为的物理桥梁,再通过机器学习实现高通量筛选,最后用SHAP可解释性把"为什么选这个分子"的机理说得清清楚楚——"描述符筛选→模型预测→机理解码→实验验证",四步形成完整闭环。
这套框架的通用性值得关注。LUMO能级描述的不只是锌电池——多价金属电池(Mg、Ca、Al)同样面临界面不稳定问题,羰基的电子局域化调控策略,原理上是相通的。这篇论文发表在Advanced Materials,编辑给了一句很到位的评价:"establishes a universal theoretical framework for rational electrolyte additive engineering in multivalent metal batteries"——为多价金属电池的电解液添加剂理性设计,建立了通用理论框架。
当然,冷静地说,从扣式电池到实用化,中间还有距离。
扣式电池里表现出色的方案,迁移到软包电池时往往面临新的工程挑战:电解液浸润均匀性、大面积锌箔的表面一致性、高温/低温极端工况……这些都需要在实际电池尺度上验证。OMAPNet的训练集是通用分子库,对电解液专属分子空间的泛化能力还需要进一步考察。此外,不同正极材料体系(MnO₂、普鲁士蓝等)对电解液配方的要求不尽相同,Ket策略能否与各种正极兼容,也需要系统验证。
说到底,这项研究最让我印象深刻的,不是187天的循环数据,而是背后的"设计哲学":不再依赖盲目的试错,而是从分子轨道能级出发,用AI找到那个结构上"命中注定"的分子,再把界面保护这件事,做成了精准的分子工程。
文献信息
• 标题:Carbonyl-Modulated Lowest Unoccupied Molecular Orbital Energy Directs Machine Learning-Assisted Screening of Electrolyte Additives Toward Ultra-Stable Zinc Metal Anodes • 期刊:Advanced Materials(材料顶刊) • 发表时间:Received 2026-02-10,Accepted 2026-06-10 • DOI:https://doi.org/10.1002/adma.73811
