 夜雨聆风
夜雨聆风inDecay :CRSIPR 编辑产物预测
好久没更新了。今天趁着一个做了三年的工作基本收尾,赶紧和大家分享一下我们的新工具——inDecay,一个用来预测 CRISPR/Cas 基因编辑产物的模型。
🎯 太长不看版
同一个位点,CRISPR/Cas 的无模板编辑会引入很多种不同的突变。产生哪种突变,其实和细胞类型、甚至细胞的状态都很有关系。已有工具因为没考虑不同细胞里 DNA 修复通路的差异,在小鼠胚胎上常常预测不准。
我们的新工具 inDecay:
特征设计更高效:在多种细胞、多个指标上都超过了已有工具。 模型足够轻:只要 30–50 个样本‼️,就能把一种细胞学到的修复偏好,迁移到另一种细胞上。这让编辑产物预测第一次能用在样本稀缺的场景,比如小鼠胚胎、大动物胚胎。 一套大规模小鼠编辑数据:共 52 个位点、超过 1,878 个胚胎、总计 11,787 条产物。 基于这套数据,我们训练了适用于小鼠胚胎 2–4 细胞期的预测模型,并揭示了小鼠胚胎独特的修复偏好。 还有猪、牛、山羊、绵羊四种大动物的胚胎编辑数据:即使只用 12 个靶点的小数据,也能带来明显的偏好迁移。
🧑💻 代码仓库:https://github.com/StatBiomed/inDecay
🌐 网页预测工具:https://indecay.online/
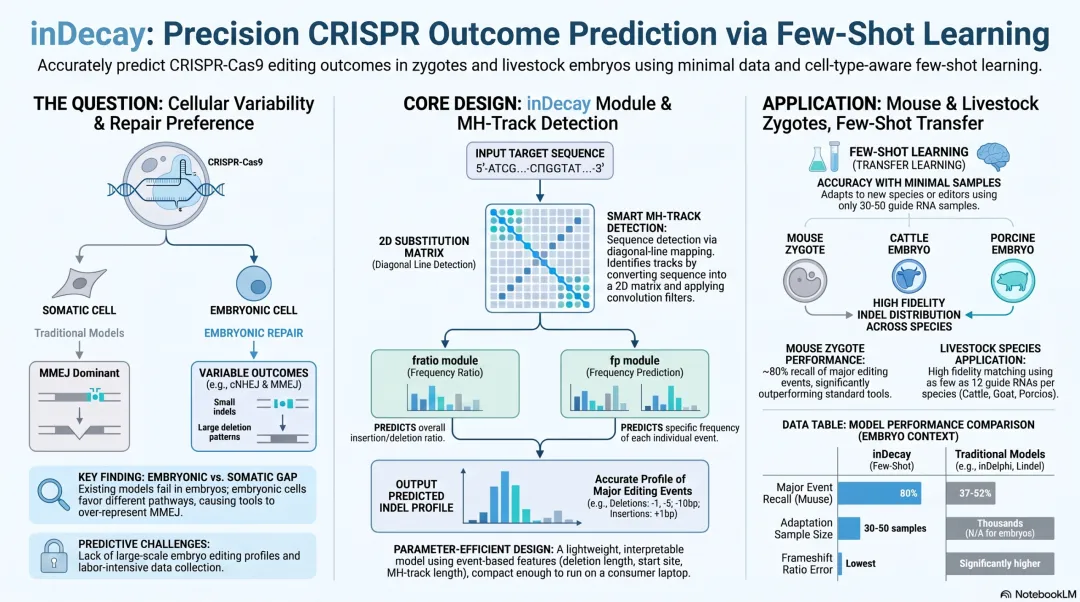
Precision CRISPR Outcome Prediction 概览图 *NotebookLM 生成
预测编辑产物,不是早就做烂了吗?
CRISPR/Cas 的预测,脱靶率、编辑活性,相关的 AI 工具早就卷上天际了,还能玩出什么新花样?
这次 inDecay 不只是预测一个数,而是预测一整张图谱。给定一条靶点序列,它能预测出这个位点所有可能的编辑产物,以及每一种增删(indel)出现的概率。
什么意思?就是说,同样一条靶点序列和 gRNA,每次编辑切出来的增删都不太一样,带点随机性——有点像抽盲盒、抽卡。
CRISPR/Cas 编辑产物到底有多复杂
下面这张图,是一次无模板编辑(template-free CRISPR/Cas editing)产生的增删分布(indel distribution)。
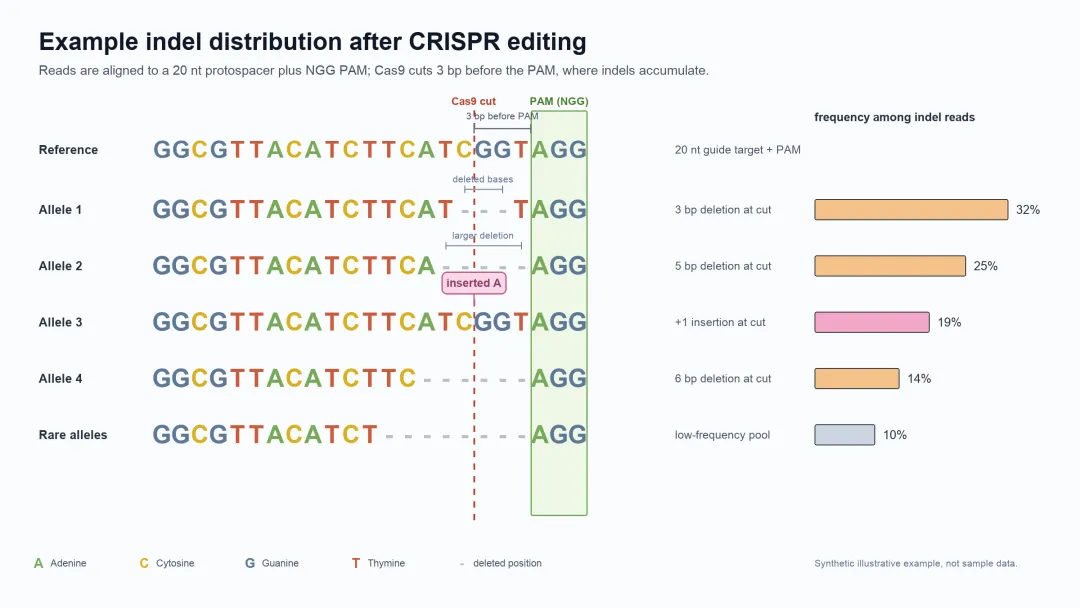
indel distribution 示意图
* 增删分布(indel distribution),文中也叫它"产物分布""修复画像"。
图里展示了五种不同的突变(一种突变对应一种 allele)。产物之所以这么多样,是因为删除(deletion)和插入(insertion)各自都有随机性:删除的长度和起始位置在变,插入的长度和碱基组合也在变,两两组合,产物数量一下就爆了。
只算最常见的 1–38 bp 删除和 1–2 bp 插入,一个靶点大约就能产生 300–400 种不同产物。更长片段的删除,出现频率和位置我们这里暂时不考虑。
决定产物的两大因素:靶点上下文 + 修复通路偏好
产物这么复杂,我们从哪儿下手?
好在,虽然有随机性,编辑产物主要还是由两件事决定:靶点的序列上下文,以及细胞对不同 DNA 修复通路的偏好。
下图是四条主要的 DNA 修复通路,它们各自产生什么突变,其实是有规律可循的。其中 HDR 需要模板,SSA 倾向于产生大片段删除——这两类都不在我们无模板编辑的系统里。所以真正要理解的,是 MMEJ 和 c-NHEJ这两条。而每条通路具体怎么反应,就得看靶点序列的上下文了。
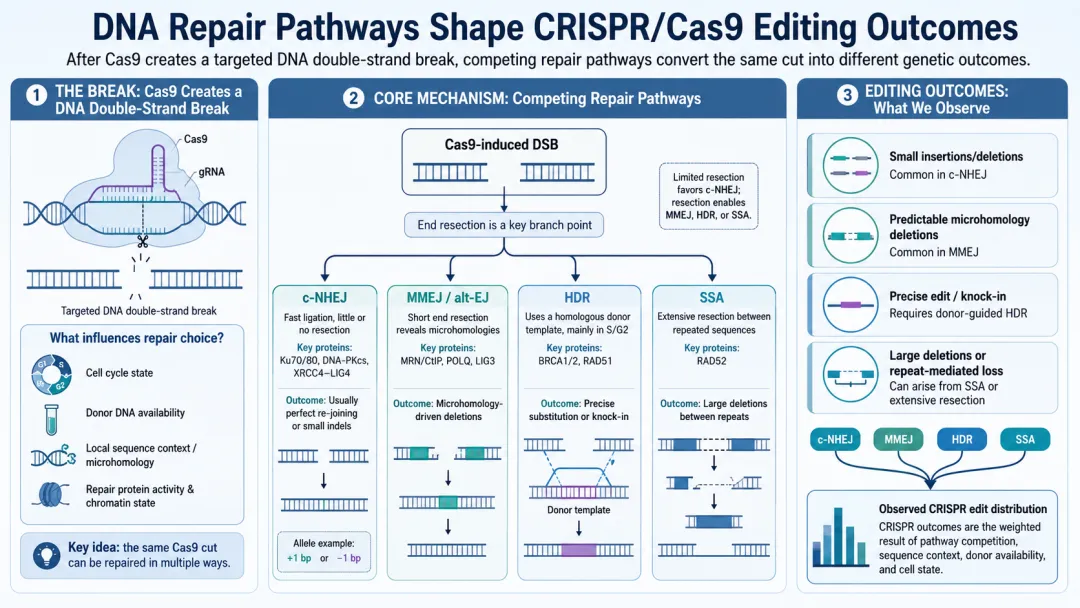
(配图:四条 DNA 修复通路示意图)
有些产物和上下文关系特别强。比如微同源末端连接(Microhomology-Mediated End Joining,MMEJ):切口两端如果有同源序列,修复时会错误修剪,就产生了对应的删除(见上图 MMEJ 部分)。所以只要我们把两端匹配的序列检测出来,再按它们的距离、匹配程度做成特征,一大类删除产物就能预测得很好了。
c-NHEJ则更多产生短插入和点突变。几项大规模编辑实验(Massively Parallel Assay of Self-Targeting Sequences)揭示了一些上下文规律:
首先,删除和插入事件的比例也受上下文序列影响。Max Shen 等人的 Nature2018(David Liu、David Gifford、Richard Sherwood 共同通讯),以及背靠背发表的 Felicity Allen、Leopold Parts 等人 2019 Nature Biotechnology,都提到了下图 c 里的序列特征(0 是切口位置):靶点越接近这类序列,插入突变的占比越高。 其次,插入进去的碱基,往往是切口附近序列的拷贝——也就是"近邻拷贝"现象。
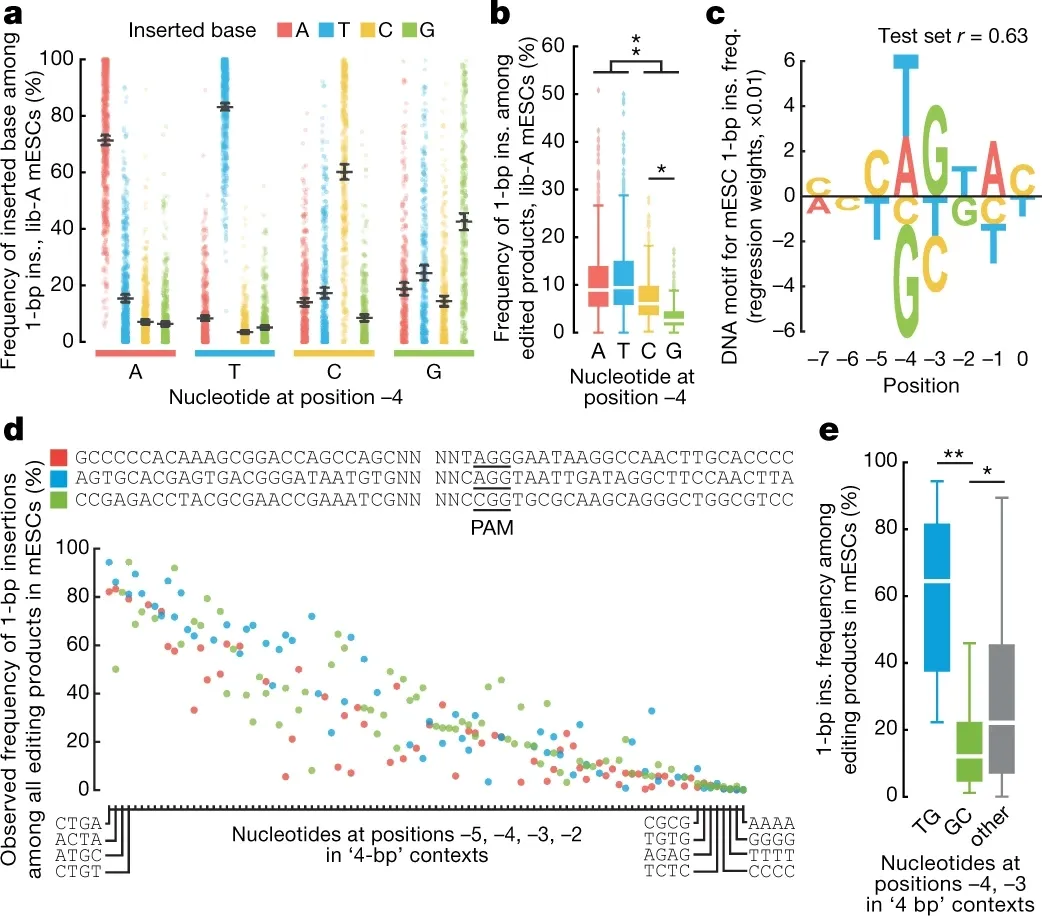
Fig 2 from Shen, M.W., Nature 2018. https://doi.org/10.1038/s41586-018-0686-x
搞清楚这些规律,我们就能设计出合理的特征了。
inDecay 的两层设计:先验特征工程 + 轻量化模型
一位专门做小鼠胚胎编辑的老师告诉我们:现有的预测软件在小鼠胚胎上经常预测不准。这大概和胚胎二细胞期修复通路的基因表达有关。后面的合作,还有 inDecay 的诞生,都是从这句话开始的。
现有模型(inDelphi、ForeCasT)都是在大规模平行实验数据上训练的。这些数据确实揭示了不少通用的上下文规律,但它们同时也"混进了"实验所用细胞类型的通路偏好——而小鼠胚胎自身的偏好,和这些已有细胞都不一样。怎么既用上通用规律,又剥掉细胞特异性,就成了整个课题的关键。
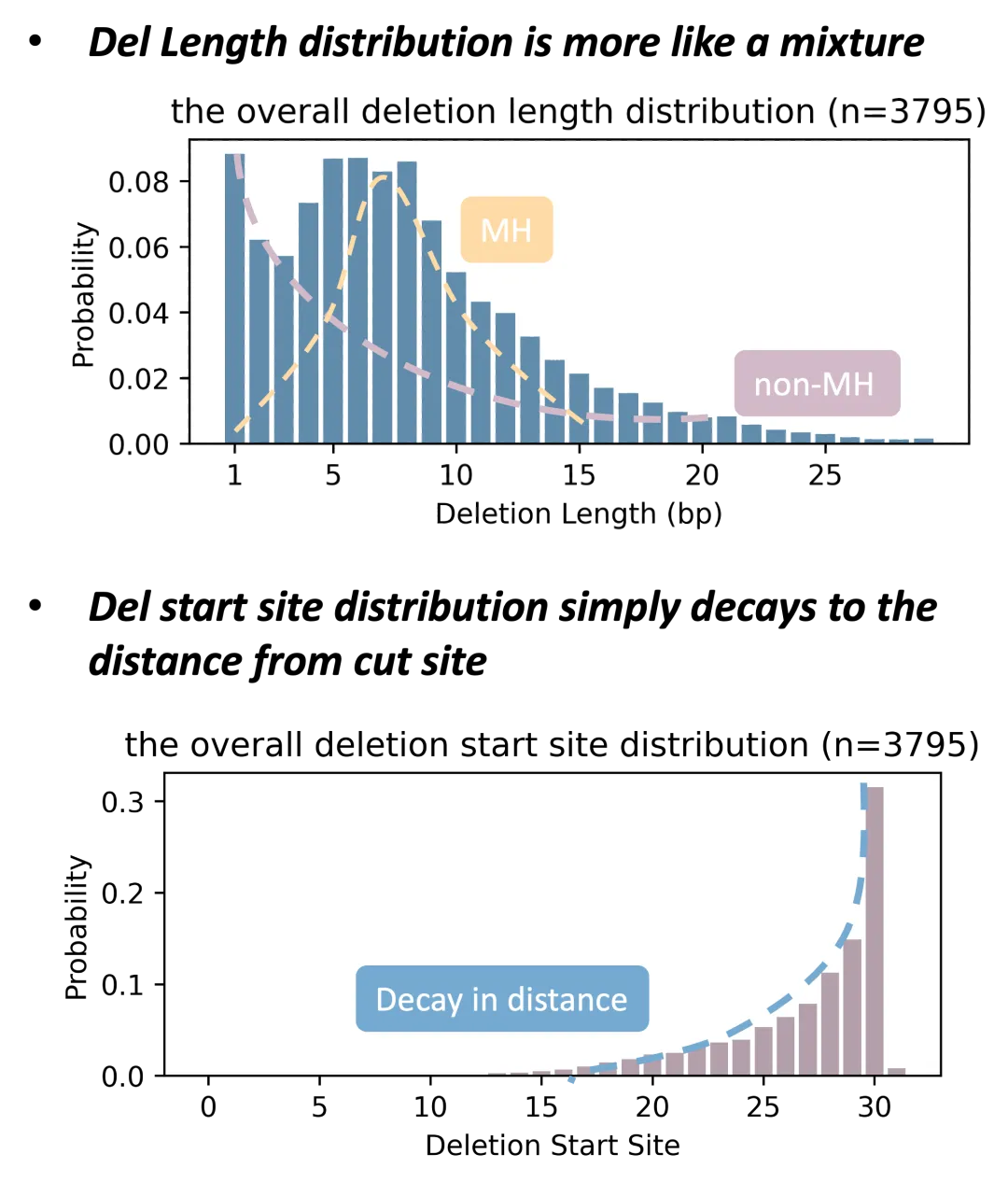
dss-frequency 图
在 Lindel 数据上做初期分析时,我们发现:删除事件的概率,和它的起始位置(start site,紫色)强烈负相关;而在删除长度上(deletion length,蓝色),却出现了双峰。后来我们按有没有微同源序列,把事件分成两类——有 MH 的走 MMEJ 通路,没 MH 的走 c-NHEJ 通路——趋势立刻就清楚了:没有 MH 时,删得越长概率越低;一旦有 MH,对应长度的概率会被拉高,双峰就是这么来的。
于是,基于修复机制的先验知识,加上数据的初步统计,我们提炼出了三个核心特征。后来又陆续补上了邻近碱基等特征。而增删(indel)建模的核心,就是这两条衰退(decay)规律——inDecay 这个名字也是这么来的(indel prediction by frequency Decay)。
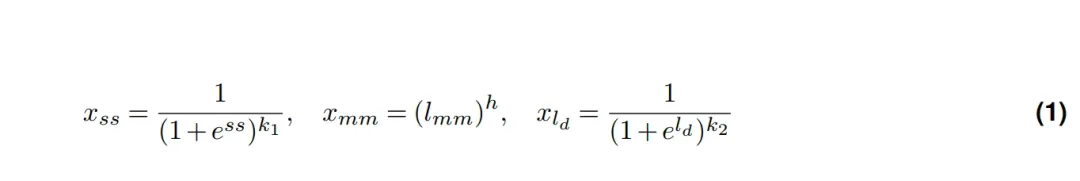
inDecay 特征示意图
有了这些高度浓缩的特征,还差最后一块:细胞通路偏好的建模。可惜的是,我们目前只能在拿到编辑数据之后,统计每种细胞的通路偏好(见正文 Fig S2),还没法光凭细胞的某些特质就提前算出来,只能事后建模(post-hoc)。
模型表现,通常由两件事共同决定:特征的表达能力和模型的参数量。我们的思路是:把特征做到极致,让模型不用再花参数去"提取信息",省下来的参数就能专门用来学细胞的通路偏好。这样一来,只用很少的参数,就能抓住不同细胞的特异性——也为后面小鼠胚胎这种稀缺数据的拟合打好了底子。(说白了就是:数据少,变量就得少,方程才有解。)
模型的大思路就是这样。更细的部分等文章正式出来再展开。感兴趣的同学可以先看预印本 🙇♂️.
代码仓库:https://github.com/StatBiomed/inDecay
想直接试试?网页工具:https://indecay.online/ 预印本 :查看原文👇 或访问 https://www.semanticscholar.org/paper/Accurate-prediction-of-CRISPR-editing-outcomes-in-Zheng-Yu/1d6959eb20ab5c252e679feccbfbc987685d086a
实际应用:用 Sanger 数据建模小鼠和大动物胚胎编辑
轻量化模型最大的好处,就是好迁移。在各个大数据集上预训练之后,我们发现,只要大约 30–50 个样本(靶点),就能把关键指标提升到合理水平。
这里要特别感谢合作者提供的大量胚胎编辑数据。其中最有价值的一步,是我们打通了用 Sanger 一代测序数据反推增删分布的流程。此前的数据几乎都来自 NGS 二代测序,需要专门的实验平台,全世界只有少数实验室能产出。但 Sanger 测序,恰恰是很多实验室最日常的手段——不少实验室其实攒了很多年的 Sanger 数据。用我们的处理流程,每个实验室都能给自己的细胞系或动物模型,搭一个专属的编辑预测模型 🤩。
为了更好地服务小鼠基因编辑品系的构建,合作者收集了 52 个基因组位点、1,878 个胚胎、11,787 条突变的数据。小鼠胚胎还得靠人工操作来获取和引导突变,这背后是好几位技术员和硕博接力,付出了大量人力物力。没有这些珍贵的数据,算法本身也谈不上什么价值。在共同一作 Louisa 的努力下,我们把胚胎数据处理好,并在上面调出了最优的小鼠模型(Louisa 的耐心和细致 💪)。
最后的结果:inDecay 在小鼠数据上能预测出 80% 的主要事件,在总体分布、移码突变概率等关键指标上,都大幅领先已有工具。这其实也不奇怪——现有模型的训练数据和小鼠胚胎差得太远,而 inDecay 只要能从小样本里学到鼠胚的通路偏好,就能在各项指标上超过同行
 。
。

小鼠模型效果对比图
数据处理的血与泪
小鼠和大动物的胚胎编辑数据太珍贵,每一条 Sanger 数据都得仔细分析。而这类数据不像 NGS,没有那么多现成工具能直接调用。
第一个坎就是:Sanger 的 ab1 数据不是纯文本,而是信号的叠加。如果一个位点有多个等位突变,信号就是混在一起的,得先把它拆开。这一步 Louisa 做了大量分析和比较,摸清了 DecodR 和 Synthego 各自的优劣,最终定下用 DecodR。我前期写的一堆分析代码,也被她重构得更干净、更好复用。Respect!
当然,也少不了各位合作者和老师们的悉心指导 🙏。
快来试试
最后再给工具打个广告 👉 https://indecay.online/
把靶点序列输进去,填上切口位置(PAM 前三位)和最接近的细胞类型,就能预测出所有可能的产物和它们的概率了。
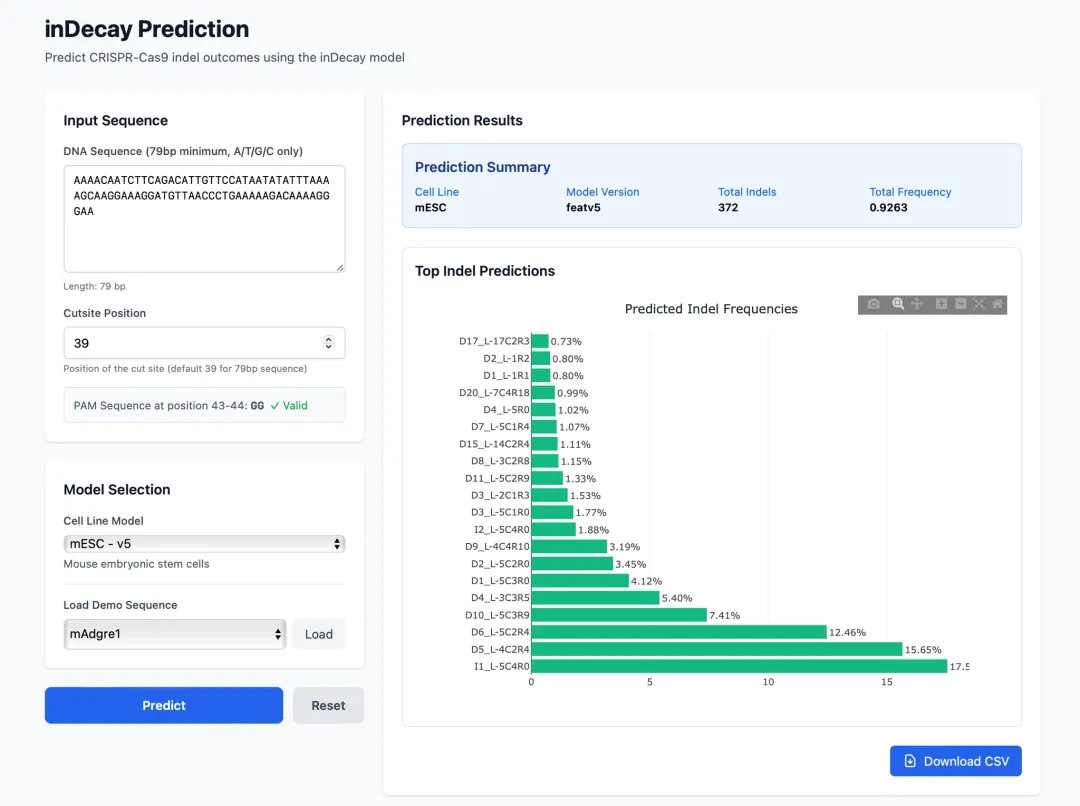
结果还能一键下载,打开就能看到对应的序列和概率。
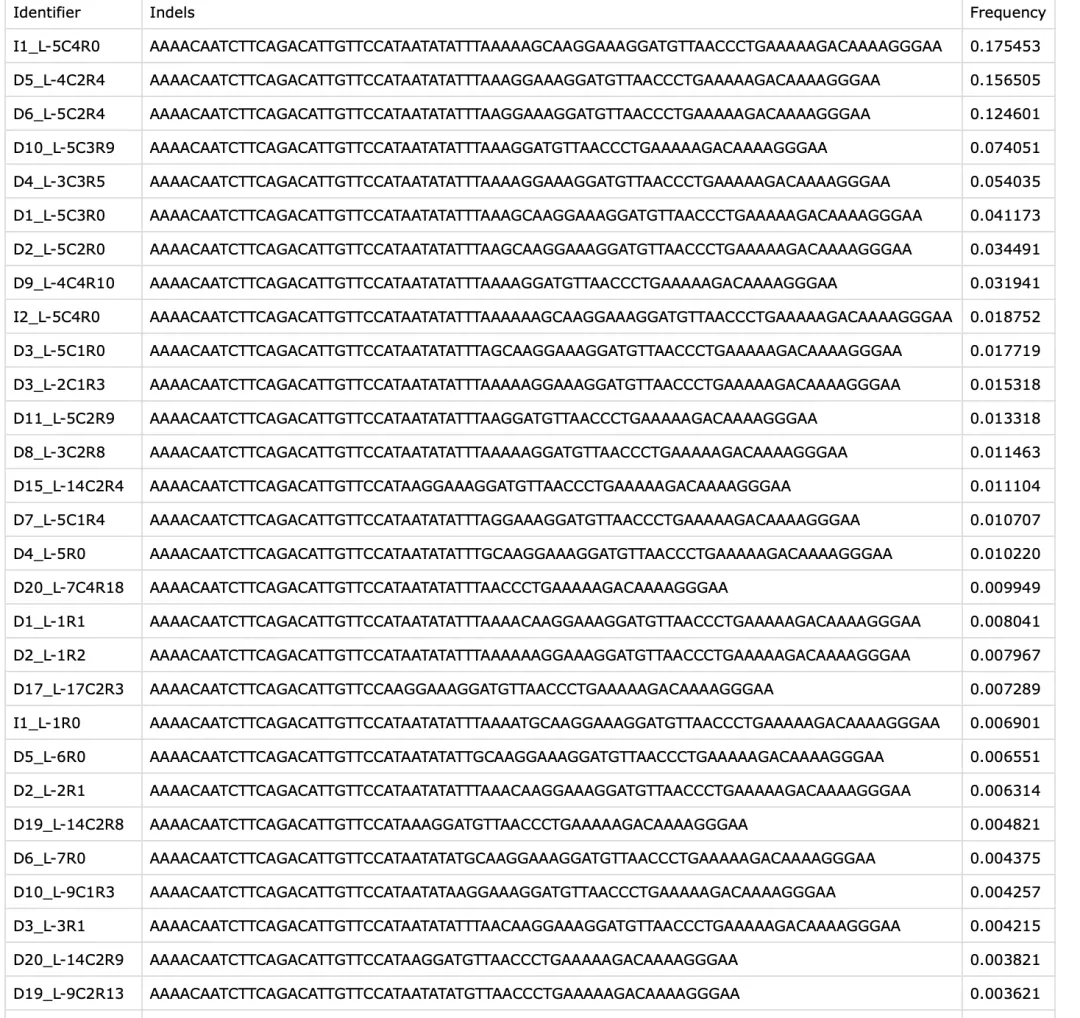
预测结果下载界面

