夜雨聆风
夜雨聆风





使用生信软件,必须阅读文档
很多人使用生物软件,拿过来就用,不看文档。这是非常不好的习惯,而且有可能酿成大错,比如将原核基因预测的软件预测真核生物基因,这是万万不行的。有时候一个选项参数设置错误,就会造成错误的结果。学习生物信息软件,学会如何阅读软件文档非常重要,本文介绍生物软件如何查看文档。
什么是生物软件?
生物信息本质上是使用计算机软件来处理测序数据,所以正确使用生物软件很重要。软件的定义是程序加文档两部分,软件=程序+文档,我们主要是运行程序,很多人忽略软件的文档部分。所谓软件文档就是软件的说明书,这要介绍如何使用软件。文档可以包括软件介绍,作者,版本,使用说明,常见问题,联系方式等内容。很多人常常忽略文档部分,其实文档是软件不可或缺的一部分,相当于软件的一份说明书,熟读文档之后,就可以熟练使用软件了。
如何查看文档?
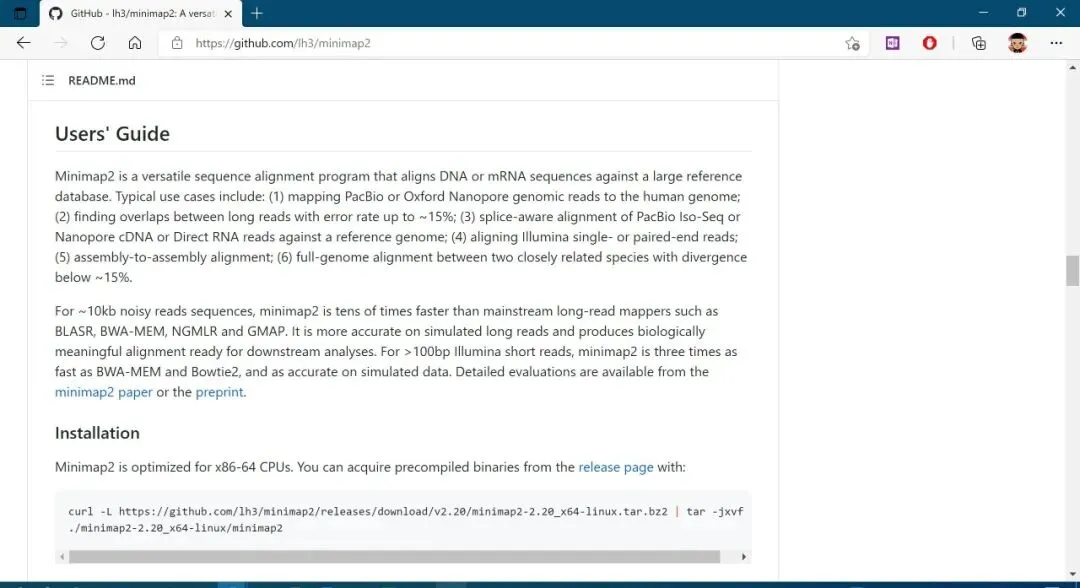
其实每款软件都需要带文档,只不过不同软件文档书写水平不同,一款好的软件必须有详细的说明文档。对于windows图形化软件,可以使用F1快捷键直接调用文档。而很多生物软件采用命令行模式,可以通过以下方式查看文档:直接命令行输入程序名,不加选项参数;也可以直接添加“–help”选项,有些软件是-h选项或者-help,不同软件有所差别,例如想查看minimap2帮助文档。
(base) meta 09:38:10 ~
$ minimap2 --help
Usage: minimap2 [options] <target.fa>|<target.idx> [query.fa] [...]
Options:
Indexing:
-H use homopolymer-compressed k-mer (preferrable for PacBio)
-k INT k-mer size (no larger than 28) [15]
-w INT minimizer window size [10]
-I NUM split index for every ~NUM input bases [4G]
-d FILE dump index to FILE []
Mapping:
-f FLOAT filter out top FLOAT fraction of repetitive minimizers [0.0002]
-g NUM stop chain enlongation if there are no minimizers in INT-bp [5000]
-G NUM max intron length (effective with -xsplice; changing -r) [200k]
-F NUM max fragment length (effective with -xsr or in the fragment mode) [800]
-r NUM[,NUM] chaining/alignment bandwidth and long-join bandwidth [500,20000]
-n INT minimal number of minimizers on a chain [3]
-m INT minimal chaining score (matching bases minus log gap penalty) [40]
-X skip self and dual mappings (for the all-vs-all mode)
-p FLOAT min secondary-to-primary score ratio [0.8]
-N INT retain at most INT secondary alignments [5]
Alignment:
-A INT matching score [2]
-B INT mismatch penalty [4]
-O INT[,INT] gap open penalty [4,24]
-E INT[,INT] gap extension penalty; a k-long gap costs min{O1+k*E1,O2+k*E2} [2,1]
-z INT[,INT] Z-drop score and inversion Z-drop score [400,200]
-s INT minimal peak DP alignment score [80]
-u CHAR how to find GT-AG. f:transcript strand, b:both strands, n:don't match GT-AG [n]
Input/Output:
-a output in the SAM format (PAF by default)
-o FILE output alignments to FILE [stdout]
-L write CIGAR with >65535 ops at the CG tag
-R STR SAM read group line in a format like '@RG\tID:foo\tSM:bar' []
-c output CIGAR in PAF
--cs[=STR] output the cs tag; STR is 'short' (if absent) or 'long' [none]
--MD output the MD tag
--eqx write =/X CIGAR operators
-Y use soft clipping for supplementary alignments
-t INT number of threads [3]
-K NUM minibatch size for mapping [500M]
--version show version number
Preset:
-x STR preset (always applied before other options; see minimap2.1 for details) []
- map-pb/map-ont - PacBio CLR/Nanopore vs reference mapping
- map-hifi - PacBio HiFi reads vs reference mapping
- ava-pb/ava-ont - PacBio/Nanopore read overlap
- asm5/asm10/asm20 - asm-to-ref mapping, for ~0.1/1/5% sequence divergence
- splice/splice:hq - long-read/Pacbio-CCS spliced alignment
- sr - genomic short-read mapping
See `man ./minimap2.1' for detailed description of these and other advanced command-line options.
在线文档
除了命令行文档之外,一般软件官网也会提供文档部分,目前很多软件官网都在github上,相应的也会提供文档部分。https://github.com/。
Read the Docs
ReadtheDocs(https://readthedocs.org/)网站是一个创建、托管和浏览文档的平台,可以直接连接到github,readthedocs可以提供更完整的文档内容。同时可以提供更多的案例,目前越来越多的软件开始将文档转到该平台。