 夜雨聆风
夜雨聆风





医疗器械软件设计开发流程-来源(2022年第9号)医疗器械软件注册审查指导原则(2022年修订版)的总结
三、详细总结
1、指导原则基础信息
- 目的:规范医疗器械软件生存周期管理、注册申报资料准备及技术审评要求,为体系核查提供参考。
- 定位:数字医疗指导原则体系的基础文件,为通用要求,注册申请人可根据产品特性调整适用性并说明理由。
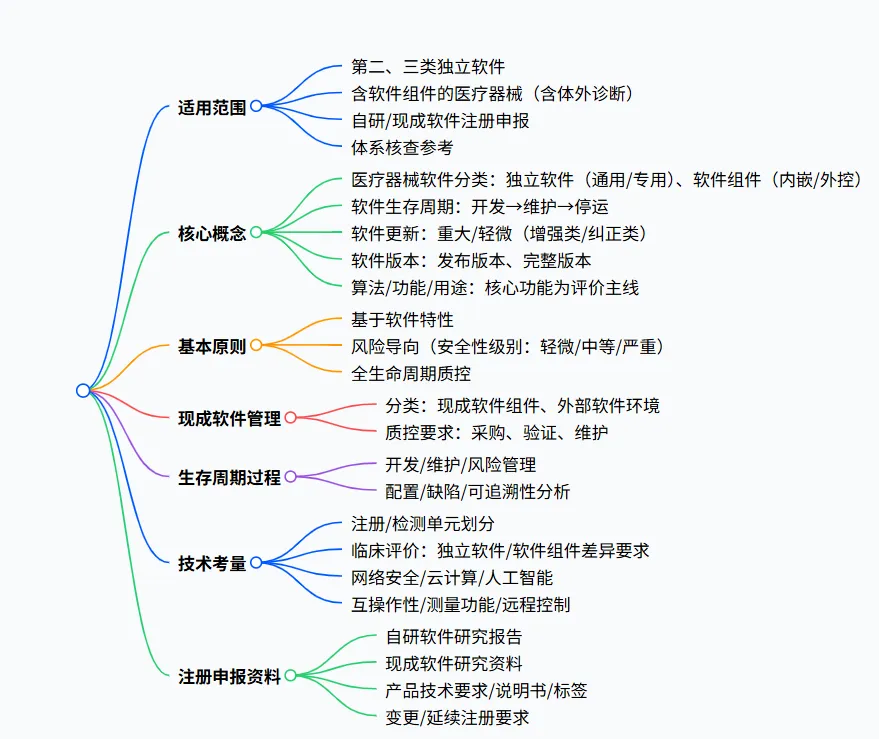
- 适用范围
-
注册申报对象:第二、三类独立软件、含有软件组件的医疗器械(含体外诊断医疗器械);自研软件、现成软件均适用。 -
其他用途:医疗器械软件、质量管理软件的体系核查参考。
2、核心概念界定
|
|
|
|---|---|
| 医疗器械软件分类 |
|
| 软件生存周期 |
|
| 软件更新类型 |
|
| 软件版本管理 |
|
| 软件安全性级别 |
|
3、三大基本原则
- 基于软件特性
-
软件无物理实体,缺陷具有固有性、更新具有快速性,需结合风险管理、质量管理和软件工程保证安全有效性。 -
软件与硬件存在本质差异,传统硬件质控方法不适用。 - 风险导向
-
安全性级别决定生存周期质控和申报资料详尽程度,严重级别对应最严格要求。 -
风险管理需贯穿全程,关注软件失效引发的危险情况,软件组件需与所属医疗器械整体开展风险评估。 - 全生命周期质控
-
上市前:完成软件验证与确认,降低可预见风险至可接受水平。 -
上市后:开展不良事件监测、软件更新质控,停运阶段需考虑数据迁移、隐私保护。
4、现成软件管理要求
- 分类
-
现成软件组件:作为医疗器械软件组成部分(遗留 / 成品 / 外包软件),属于监管对象。 -
外部软件环境:系统软件、通用应用软件等,非监管对象,但需评估对医疗器械软件的影响。 - 质控要点
-
采购阶段:评估现成软件的成熟度、兼容性、售后支持。 -
开发阶段:开展差距分析,必要时采取补救措施。 -
维护阶段:外部软件环境更新若跨越不兼容平台,属于重大软件更新。
5、软件生存周期过程
- 核心过程:软件开发、软件维护、软件风险管理、软件配置管理、软件缺陷管理,可追溯性分析需贯穿全程。
- 敏捷开发要求:需兼顾质量管理体系,重点控制软件更新、文件与记录。
- 关键活动
-
验证:通过单元 / 集成 / 系统测试等证明输出满足输入要求。 -
确认:通过用户测试、临床评价证明满足用户需求和预期用途。
6、技术考量重点
- 注册与检测单元划分
-
独立软件:按管理类别、预期用途、功能模块划分;功能复杂的需拆分(如 PACS 拆分为平台软件 + 特定功能软件)。 -
软件组件:与所属医疗器械同一注册单元;检测需覆盖不同核心算法对应的核心功能。 - 临床评价原则
-
独立软件:非辅助决策类基于核心功能比对同品种;辅助决策类基于核心算法比对,全新算法 / 功能需开展临床试验。 -
软件组件:控制 / 前处理功能随硬件整体评价,后处理功能参照独立软件。 - 新技术与特殊要求
-
云计算:视为现成软件,需评估云服务商资质、数据安全、迁移方案。 -
人工智能:需遵循专门指导原则,关注算法可解释性、数据质量。 -
网络安全:具备数据交换、远程访问功能的软件需满足相关要求。 -
测量功能:需明确准确性指标(线性度、精度等),图形学测量需在说明书标注警示。
7、注册申报资料要求
- 核心资料
-
自研软件:提交研究报告(含基本信息、实现过程、核心功能、结论),内容详尽程度与安全性级别匹配。 -
现成软件:组件需提交研究资料,外部环境需提交评估报告。 - 产品技术要求
-
独立软件:明确版本命名规则、性能指标(功能、接口、运行环境等)、附录图示(体系结构、物理拓扑等)。 -
软件组件:在所属医疗器械技术要求中规范版本、功能、接口等信息。 - 变更与延续注册
-
重大更新:提交完善型更新研究报告、GB/T 25000.51 自测报告。 -
轻微更新:通过质量管理体系控制,下次注册时提交资料。 -
延续注册:通常无需提交软件资料,需在技术要求中明确版本信息。
8、其他关键要求
- 质量管理软件:非医疗器械软件,无需注册,参照医疗器械软件开展确认。
- 进口软件:需提供原产国上市证明,存在中外差异的需提交说明材料。
- 标准引用:需符合 GB 4943.1、GB 9706 系列等安全标准,遵循 YY/T 0664 等软件生命周期标准。
四、关键问题
问题 1:如何判定医疗器械软件的重大更新与轻微更新?二者的注册管理要求有何不同?
答案:判定核心在于是否影响医疗器械的安全性或有效性。
重大更新:属于影响安全有效性的增强类更新,包括核心功能 / 算法 / 预期用途改变、运行环境跨不兼容平台等情形,需申请变更注册。
-
轻微更新:不影响安全有效性的增强类更新、所有纠正类更新(缺陷修复),通过质量管理体系控制,无需单独注册,待下次变更注册时提交资料。
问题 2:医疗器械软件的安全性级别如何判定?不同级别对应的质控要求有何差异?
答案:安全性级别结合预期用途、使用场景、核心功能综合判定,分轻微、中等、严重三级,也可参考同类产品不良事件 / 召回情况判定。
- 轻微级别:不可能产生伤害,质控要求最低,如概述生存周期过程、仅提供系统测试和用户测试报告。
- 中等级别:可能产生轻微伤害,质控要求中等,如提供生存周期流程图、详述风险管理过程、列明软件剩余缺陷影响。
- 严重级别:可能导致严重伤害或死亡,质控要求最高,如提供设计历史文档集索引表、开展集成测试、提交完整的更新历史记录。
问题 3:现成软件组件与外部软件环境在监管要求上有何区别?注册申报时分别需要提交哪些资料?
答案:二者核心区别在于是否为医疗器械软件的组成部分,监管要求差异如下:
- 现成软件组件
-
性质:属于医疗器械软件组成部分,是监管对象,直接实现医疗用途。 -
申报资料:部分使用方式下,在自研软件研究报告中说明相关情况;全部使用方式下,单独提交现成软件组件研究报告(含标识、风险管理、验证确认等)及类型判定依据(如外购合同、外包协议)。 - 外部软件环境
-
性质:为医疗器械软件运行提供支持(系统软件、通用软件等),非监管对象。 -
申报资料:提交外部软件环境评估报告,内容包括安全性级别、软件标识、风险管理、验收管理、维护计划等,严重级别需额外提供兼容性测试报告和停运维护方案。
