夜雨聆风
夜雨聆风





FDA发布《用于生产和质量管理体系软件的验证指南》


信息来源:FDA,整理:医咖李弘老师
2026 年 2 月 3 日,美国食品药品监督管理局(FDA)正式发布《用于生产和质量管理体系软件的计算机软件保证指南》(最终版),该指南全面取代了 2025 年 9 月 24 日发布的初始版本,成为医疗器械行业软件合规的全新风向标。这份聚焦 “计算机软件保证”(Computer Software Assurance)的指导性文件,以风险为核心重构了软件合规逻辑,为医疗器械生产及质量管理体系中的软件应用提供了更灵活、更贴合技术发展的合规路径。

与传统软件验证要求不同,2026 版指南首次明确将 “计算机软件保证” 定义为基于风险的系统性方法—— 通过识别软件在医疗器械生产和质量管理中的实际用途与潜在风险,匹配相应的保证活动,最终建立并维持软件 “持续合规的验证状态”。
随着自动化、机器人技术、人工智能及云计算在医疗器械领域的广泛应用,传统 “一刀切” 的验证模式已难以适应快速迭代的软件生态。指南特别强调:其推荐的方法不具有法律强制性,制造商可在满足《联邦食品、药品和化妆品法案》及相关法规的前提下,采用替代方案,但需保留充分的风险论证与客观证据。这一弹性原则为行业实践留出了充足空间。
指南搭建了 “识别用途 – 风险分析 – 变更管理 – 确定活动 – 补充考量 – 记录留存” 的六步风险框架,形成闭环式软件保证流程:
1. 精准识别软件的 “预期用途”:划定合规边界
指南将医疗器械相关软件按用途分为三类,明确不同类别的合规要求:
- 直接用于生产 / 质量管理的软件
如自动化生产流程控制、质量数据采集与分析、质量记录维护等,需全面验证; - 支持生产 / 质量管理的软件
如测试脚本开发工具、生产记录辅助软件等,通常风险较低,可简化验证流程; - 非相关软件
如通用邮件、会计系统、基础网络设施等,无需遵循本指南的验证要求。
值得注意的是,对于云计算(IaaS/PaaS/SaaS)等新型部署模式,指南强调 “用途优先于形式”。例如,用于存储质量记录的 IaaS 云存储需满足严格的合规要求,而仅存储非质量相关生产数据的同类服务则可采用简化保证流程。
2. 风险分级:区分 “高过程风险” 与 “非高过程风险”
这是指南的核心操作要点。FDA 将软件风险分为两级,对应不同的保证强度:
- 高过程风险
软件失效可能导致质量问题,进而危及患者安全。典型场景包括:维持关键生产参数(温度、压力等)、自动判定产品合格性、生成患者使用说明书、自动化安全数据监控等; - 非高过程风险
软件失效仅影响流程效率或数据完整性,不会直接危害患者安全。典型场景包括:培训记录管理、投诉跟踪、非关键数据统计分析等。
指南特别指出,风险评估需聚焦软件的具体功能而非整体系统。例如,同一 ERP 系统中,自动化物料校验功能可能属于高风险,而物料订单记录功能则可能属于非高风险,需分别制定保证策略。
3. 软件变更管理:明确申报路径
针对已获批 PMA(上市前批准)或 HDE(人道主义器械豁免)的医疗器械,指南明确了软件变更的申报要求:
-
若变更可能导致安全风险(如修改关键生产参数控制逻辑),需通过 30 天通知提交 FDA; -
若变更不影响产品安全有效性(如优化数据录入界面),可纳入年度报告申报。
4. 匹配适宜的保证活动:测试方法的灵活选择
指南打破了 “必须进行全面脚本化测试” 的传统认知,允许根据风险等级选择测试方法:
- 高过程风险软件
推荐采用脚本化测试(含详细测试用例、预期结果、可追溯性要求)或脚本化与非脚本化结合的混合模式,确保测试的严谨性; - 非高过程风险软件
可采用非脚本化测试,如场景测试、错误猜测法、探索性测试等,减少合规负担。其中,探索性测试作为经验驱动的测试方法,被指南特别推荐用于快速验证软件功能。测试人员基于对软件的理解自发设计测试场景,重点排查隐藏故障与意外使用情况,尤其适合迭代频繁的软件系统。
5. 额外考量因素:优化保证效率
指南允许制造商通过以下方式降低保证活动的冗余度:
-
复用供应商已完成的验证工作,如审查软件开发商的质量体系认证、开发流程文档等; -
利用现有质量体系控制措施,如关键参数监控、产品出厂检验等,抵消部分软件失效风险; -
采用自动化工具(如缺陷跟踪系统、需求追溯工具)提升保证活动效率; -
对软件供应商进行风险分级评估,结合现场审计、资质审查、数据完整性验证等多维度信息。
6. 建立合规记录:明确文档要求
指南倡导 “必要且充分” 的记录原则,软件合规记录应包含:
-
软件的预期用途与风险分析结果; -
保证活动描述(测试方法、范围等); -
发现的问题及解决方案; -
测试人员、日期及必要的审批签名。 -
数字化记录(如系统日志、审计追踪、自动化测试报告)。
指南附录提供了四个针对性案例,为不同类型软件的合规实践提供参考:
1. 不符合项管理系统
-
高风险功能:产品召回 / 纠正功能(失效可能导致不安全产品流通),需采用详细脚本化测试; -
非高风险功能:不符合项发起流程(仅影响效率),采用探索性测试即可满足要求。
2. 学习管理系统(LMS)
用户访问控制、培训记录管理等功能均属于非高风险,可通过错误猜测法(如尝试越权访问、删除审计记录)验证系统安全性,无需复杂测试方案。
3. 商业智能应用
-
高风险功能:数据连接与完整性校验(失效可能导致质量趋势误判),需严格脚本化测试; -
非高风险功能:数据可视化报表生成(仅影响数据呈现),复用供应商验证结果即可。
4. SaaS 模式产品生命周期管理系统(PLM)
其合规重点在于:
-
供应商评估(含资质、网络安全措施、数据存储安全等); -
风险管理; -
需求分析; -
设计说明; -
出厂测试; -
验收测试; -
电子签名测试; -
运行维护。
FDA 在指南中明确,该文件将与 ISO 13485:2016 及 21 CFR Part 820 等法规协同实施,制造商需结合自身产品特性构建个性化的软件保证体系。随着数字化转型的深入,软件已成为医疗器械质量安全的核心载体,2026 版指南表明FDA更加重视软件在医疗器械产品实现中的作用,唯有以风险为锚,才能在保障患者安全的前提下,充分释放软件的价值。
为感谢大家的支持,医咖特地为大家准备了价值266元的医咖网上课堂抵扣券,点击本文左下角“阅读原文”即可领取。领券后可免费购买任意一节课程并免费学习,也可使用优惠券加入医咖网上课堂一年VIP会员,原价599元,券后仅需333元!方便大家使用医咖网上课堂,找到自己需要的课程,我们为大家精心整理了课程列表,大家也可在医咖网上课堂主页的搜索框里直接搜索课程名称。
欢迎长按下方二维码进入医咖网上课堂逛逛!

点击可看大图
添加韩老师微信可获得原图
系统化学习医疗器械知识
快速提升职场核心竞争力
为感谢大家的支持,医咖特地为大家准备了价值233元的医咖网上课堂抵扣券,点击本文左下角“阅读原文”即可领取。使用券后加入医咖网上课堂一年VIP会员仅需399元,本券三天有效,过期作废!
医咖网上课堂:囊括医疗器械研发、注册、体系、临床、有源、检测及职业发展系列课堂,原价599元。点击本文左下角阅读原文可领取网上课堂优惠券266元,领取后自动跳转至医咖网上课堂主页,点击会员,选择1年VIP,自动抵扣后实际仅需支付333元即可在一年内免费学习所有网上课堂已经开设的课程和未来一年即将开设的课程,还可免费加入医咖微信群和在线打卡学习营,获得学习培训证书。
医咖知识星球数据库:囊括各种医疗器械资料6000余份,手机电脑均可随时阅读和下载。原价258元。添加韩老师微信即可以208元一年的优惠价加入。

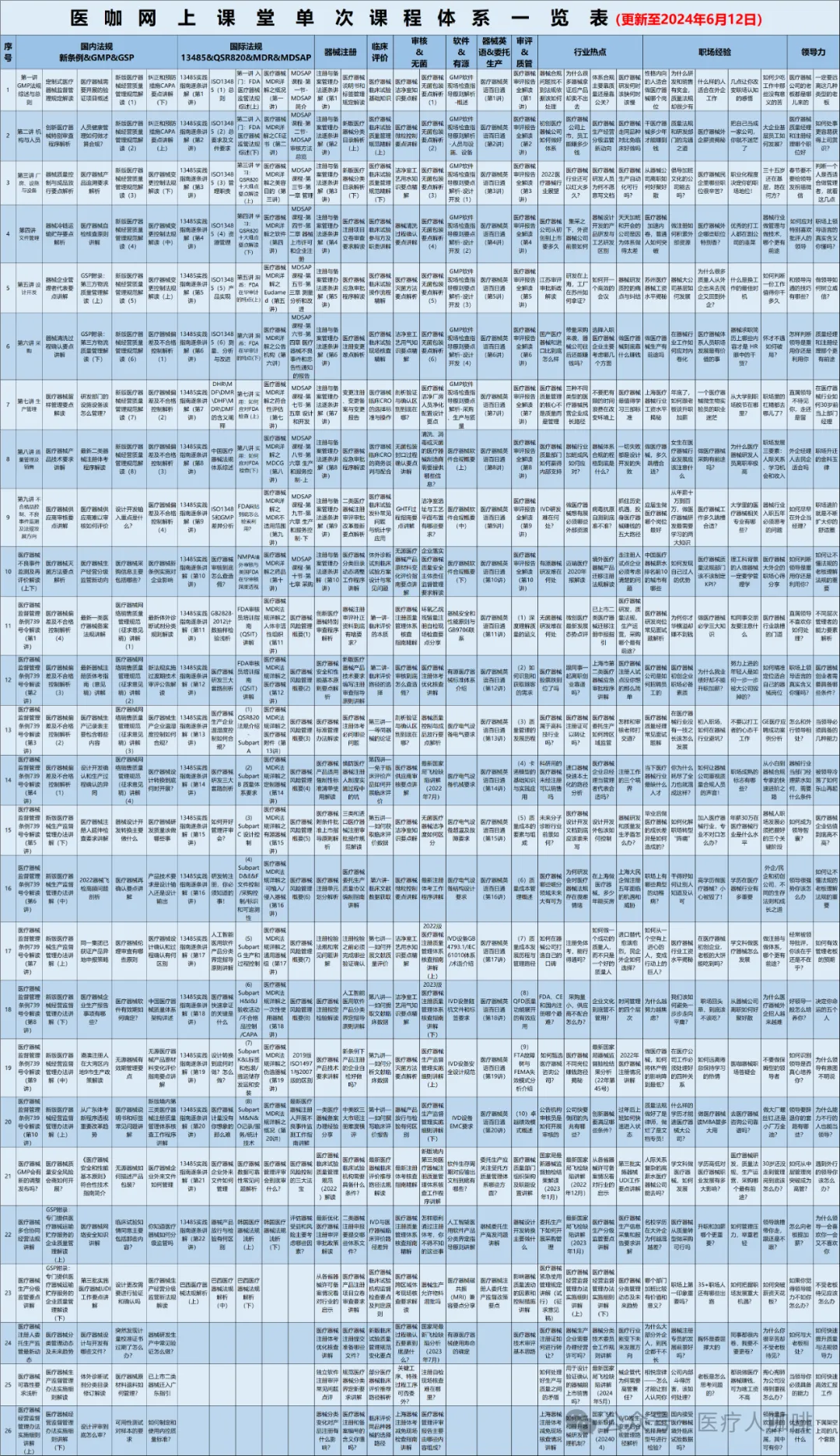
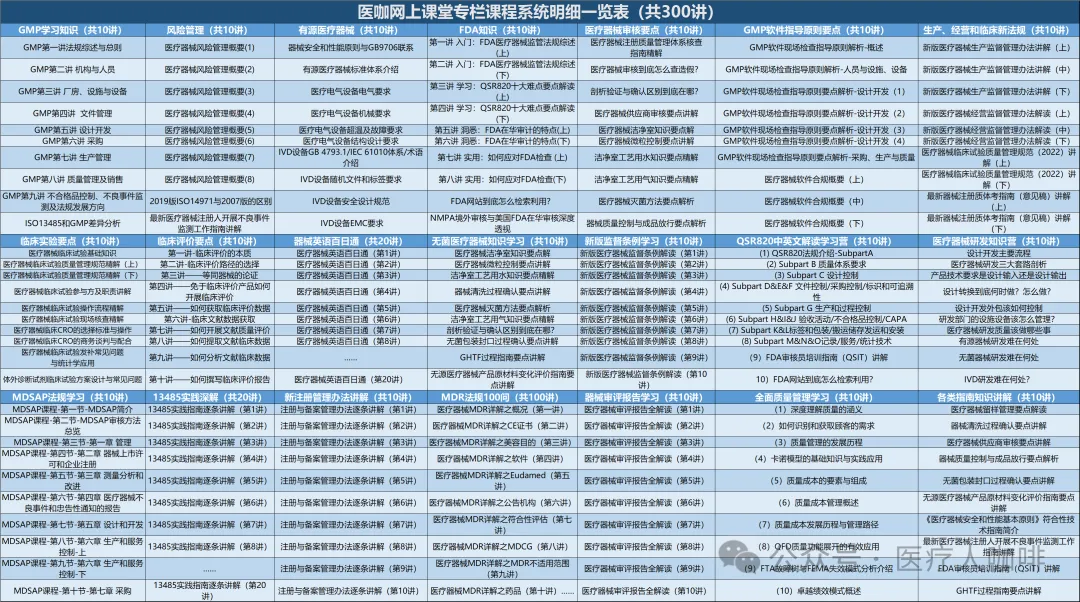
我们开设医咖网上课堂的初心就是为了方便大家学习,特别是利用零碎的时间利于上下班路上,饭后睡前的时间,能够随时随地系统性地学习医疗器械法规知识。医咖网上课堂开播四年多来,先后开设了GMP体系、13485、FDA、无菌、有源、注册、软件合规、器械英语、审评报告、全面质量管理、职场等800多节课程,深受业界欢迎!
为了帮助大家更系统、更科学、更快捷地学习我们对课程体系进行了梳理和归并,形成了近20个学习营课程体系。每个学习营课程体系涵盖10节课!
初学者只需要从系列课中选择相应的学习营,就可以系统化学习相关知识!既方便、又高效!我们强烈建议大家加入医咖网课会员,仅需333元会员费就可以免费学习网上课堂已经开设地所有课程以及未来一年新增的所有课程。同时,对于非会员,医咖也为系列课程设定了一口价,每个系列课程仅需99.99元,购买后永久有效!

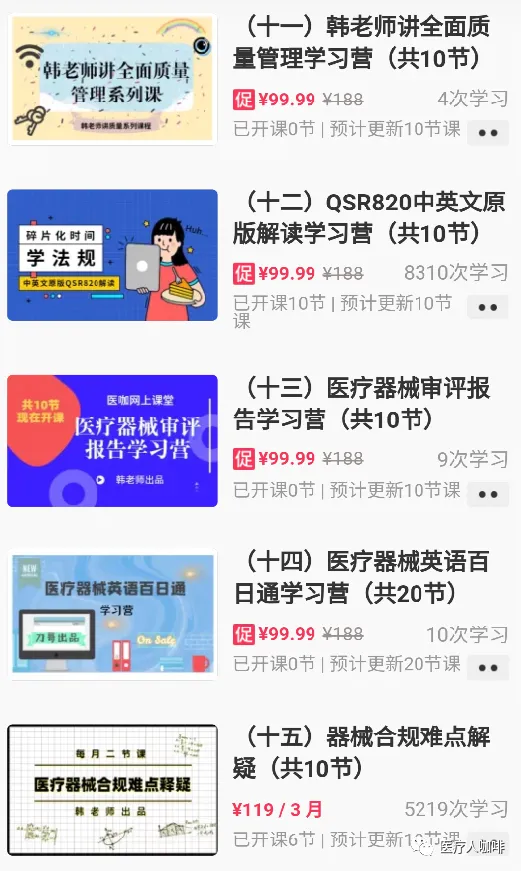
医咖网上课堂学习有何优势,我们简单列几条供大家参考:
-
所有课程配套课件及音频,课件和讲解随动播放;
-
播放速度可调,从0.8-2倍,听不懂可慢放,快放则可节省时间;
-
听到一半有事走开,课程可随时暂停,回来后记忆位置可继续播放;
-
课程内容深入浅出,语言通俗易懂,保证能听懂;
-
一个人学太孤独,加入打卡学习群,有浓厚的学习氛围,还可讨论;
-
课代表每天督促按时打卡,坚持完成打卡可获得培训证书;
-
使用千聊APP,听课不影响使用微信,适合上下班路上或开车时听

如何加入医咖网上课堂会员?
1. 点击本文左下角阅读原文即可领取200元优惠券。领取后自动跳转医咖网上课堂主页。

2. 进入医咖网上课堂主页后点击会员
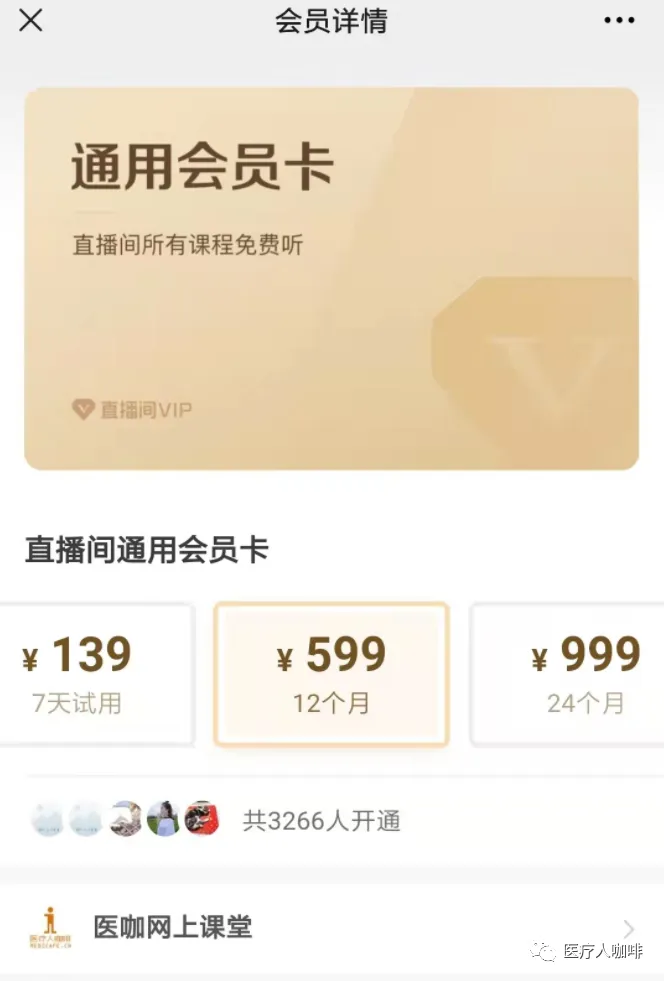
3. 选择一年VIP,原价599元,支付时自动抵扣200元,实际支付399元!
4. 成功加入会员后可添加李老师微信加入医疗咖会员群/学习营。
李老师微信二维码如下,添加微信时请注明身份