 夜雨聆风
夜雨聆风





R包神器 | ape (七) 基本和高级功能 – 分枝及进化树的注释

3.3 将数据反映到分枝上
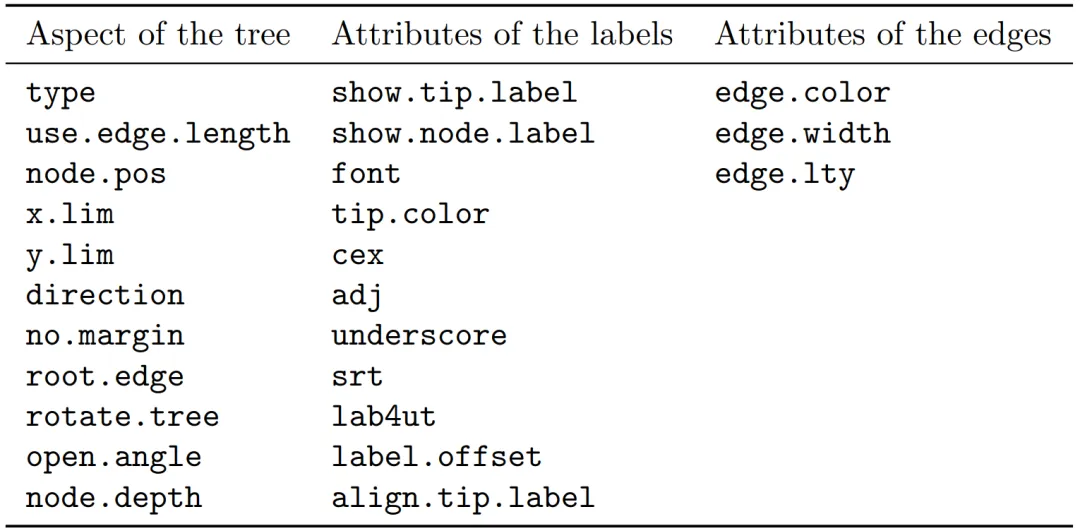
上表第3列中的3个选项控制进化树分枝的外观。与尖端的选项一样,参数也是循环使用的(Recycled)。作为参数给定的值的顺序必须与树的边的矩阵中的顺序相同。有几种方法可以找到这些数字。函数which.edge返回指定尖端标签列表的边的矩阵的行的索引,以便边(Edges)定义尖端(Tips)的分枝(Clade)。然后,可以使用返回的索引更改这些分枝的外观:
mytr$tip.label # Tip/尖端与Edge/边的概念不同:此处尖端是3个,边是4个# [1] "Pan""Homo""Gorilla"(i <- which.edge(mytr, c("Homo", "Pan")))i# [1] 2 3(i.tmp <- which.edge(mytr, c("Pan", "Homo")))i.tmp# [1] 2 3# 因此:which.edge函数中"group"参数只是集合,无顺序含义# 上面的"i"只能用于索引"边/Edge",不能用于索引"mytr$tip.labe" (且顺序与进化树尖端顺序不同,可能相反)mytr$tip.label[i]# [1] "Homo""Gorilla"# Nedge函数计算进化树中的所有的边 (x轴方向,含内部的边)co <- rep("black", Nedge(mytr))co# [1] "black""black""black""black"co[i] <- c("blue","red")co# [1] "black""blue""blue""black"par(xpd = TRUE)plot(mytr, edge.col = co)edgelabels() # 显示边(而不是尖端)的标签

扫码、联系客服老师报名,领取资料、上手分析
4 进化树的注释
一旦绘制了树,就可以使用R中的低级绘图函数 (基础绘图包 :低级绘图函数和高级绘图函数。低级绘图函数与创建新图形的高级绘图函数不同,这些函数只是将图形元素添加到现有的图形中) 添加图形元素 (Graphical elements):文本/text()、线段/segments()、点/points()、箭头/arrows()、矩形、多边形 (都在graphics包中)。这些函数需要给出绘制这些元素的坐标,这在绘制树时可能有点困难,因为默认情况下不会绘制轴/Axes(即看不到坐标)。在实际操作上,函数locator在这里可能有用。

ape有5个专门的函数也可以帮助完成这项任务:其中3个在树上添加元素,另外2个在树旁添加元素。此外,函数axisPhylo和add.scale.bar显示树的分枝的标尺/缩放/比例尺(Scale)信息。
4.1 标尺/缩放/比例尺(Scale)
函数axisPhylo绘制时间轴(A time axis),假设尖端表示当前时间,时间向后缩放(Scales backward)到根。对于非超度量(Non-ultrametric)树,取离根最远的尖端代表当前时间 (The most distant tip from the root is taken to represent present time)。这里有几个选项:
args(axisPhylo)#function (side = 1, root.time = NULL, backward = TRUE, ...)# NULL
选项backward和root.time更改scale的设置,side的含义与axis()中的相同,额外的参数(‘…’)将传递给text()。
add.scale.bar默认在图形的左下角绘制分枝长度的比例尺(Scale bar)。下面的几个选项控制其位置、格式:
args(add.scale.bar)#function (x, y, length = NULL, ask = FALSE, lwd = 1, lcol = "black",# ...)# NULL
如果ask = TRUE,函数将调用locator()识别图中的点的坐标,并要求用户单击图形,来指定绘制比例尺的位置。
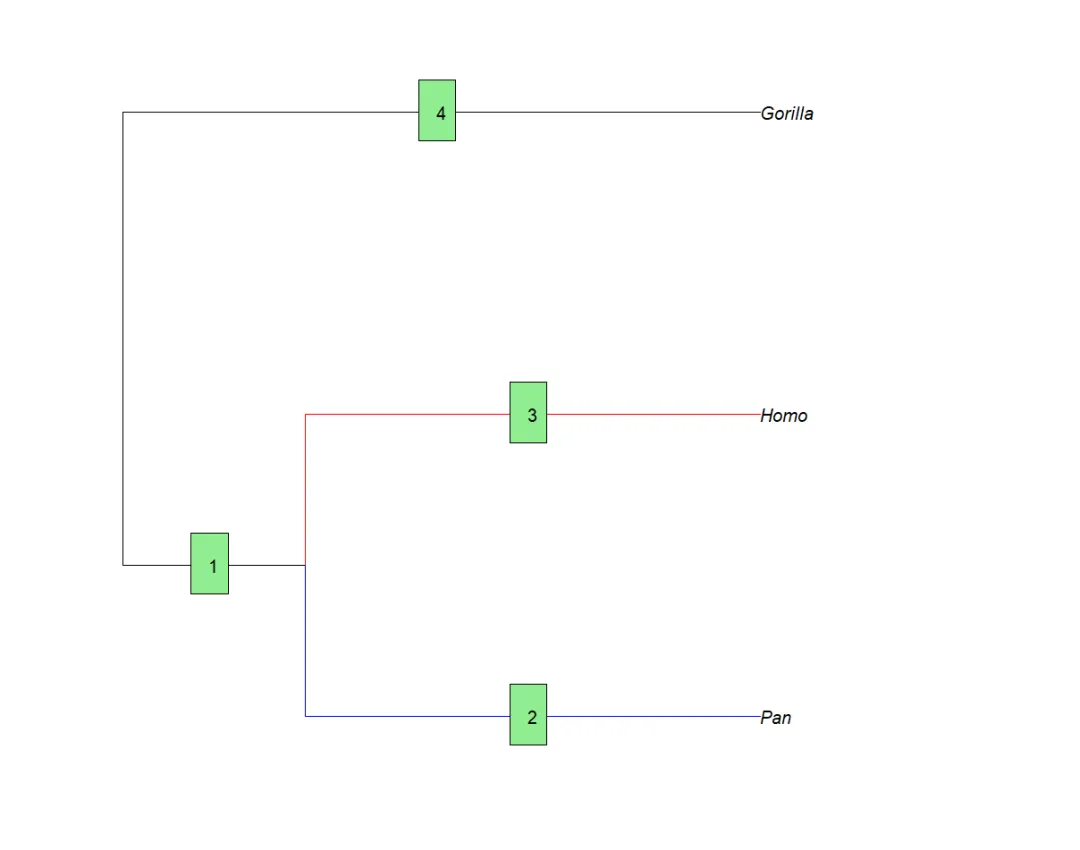
下面的示例演示了这2个函数,并在树的上方绘制了标准x轴:
plot(mytr)add.scale.bar() # 在左下角显示比例尺axisPhylo() # 在下方绘制时间轴axis(3) # 在上面显示x轴
4.2 尖端/Tips、节点/Nodes、边/Edges
nodelabels、tiplabels和edgelabels这3个函数具有几乎相同的选项,可在所绘制的进化树的节点、尖端或边上,添加注释:
args(nodelabels)#function (text, node, adj = c(0.5, 0.5), frame = "rect", pch = NULL,# thermo = NULL, pie = NULL, piecol = NULL, col = "black",#bg = "lightblue", horiz = FALSE, width = NULL, height = NULL,# ...)# NULLargs(tiplabels)#function (text, tip, adj = c(0.5, 0.5), frame = "rect", pch = NULL,# thermo = NULL, pie = NULL, piecol = NULL, col = "black",#bg = "yellow", horiz = FALSE, width = NULL, height = NULL,# offset = 0, ...)# NULLargs(edgelabels)#function (text, edge, adj = c(0.5, 0.5), frame = "rect", pch = NULL,# thermo = NULL, pie = NULL, piecol = NULL, col = "black",#bg = "lightgreen", horiz = FALSE, width = NULL, height = NULL,# date = NULL, ...)# NULL
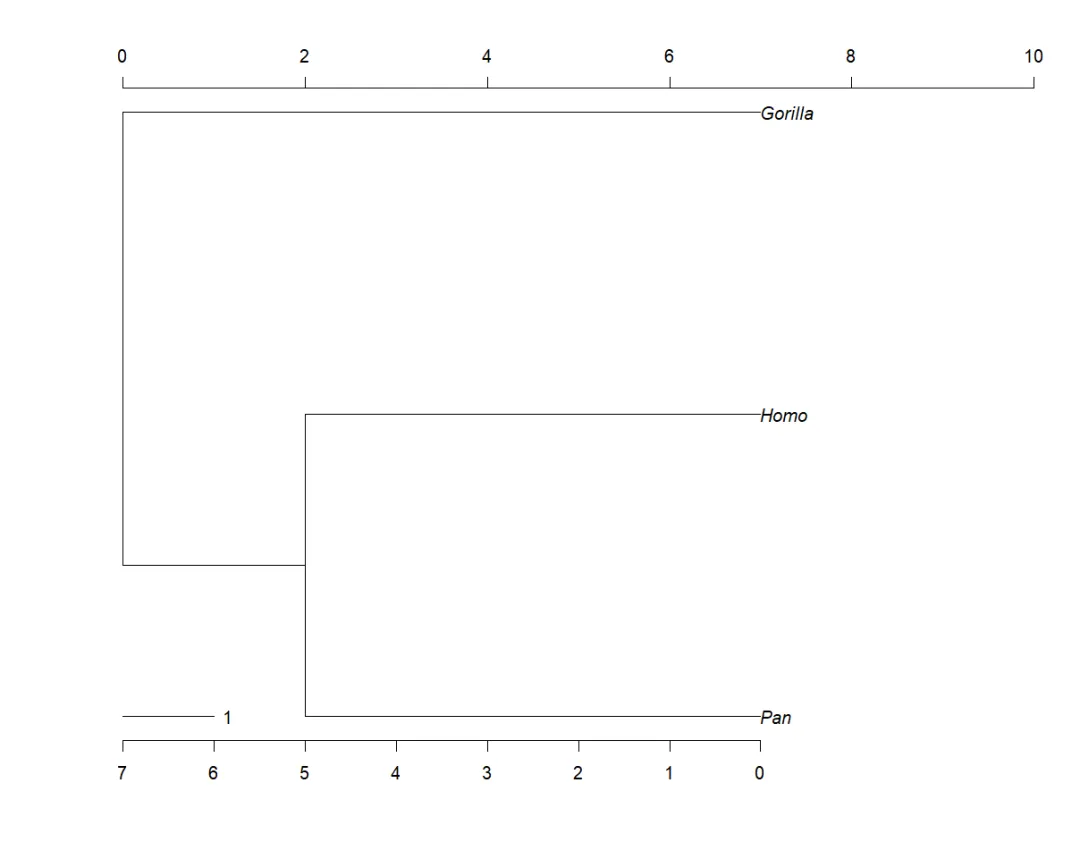
这些函数在第2个参数中(node/tip/edge)有所不同,该参数指定应打印或绘制注释的位置。如果在无任何参数的情况下使用这些函数,它们将分别打印“phylo”对象 (ape网站http://ape-package.ird.fr/ape_development.html上描述了“phylo”类对象的结构,见文末附录) 中所使用的节点、尖端或边的编号:
par(xpd = TRUE)plot(mytr)nodelabels()tiplabels()edgelabels()
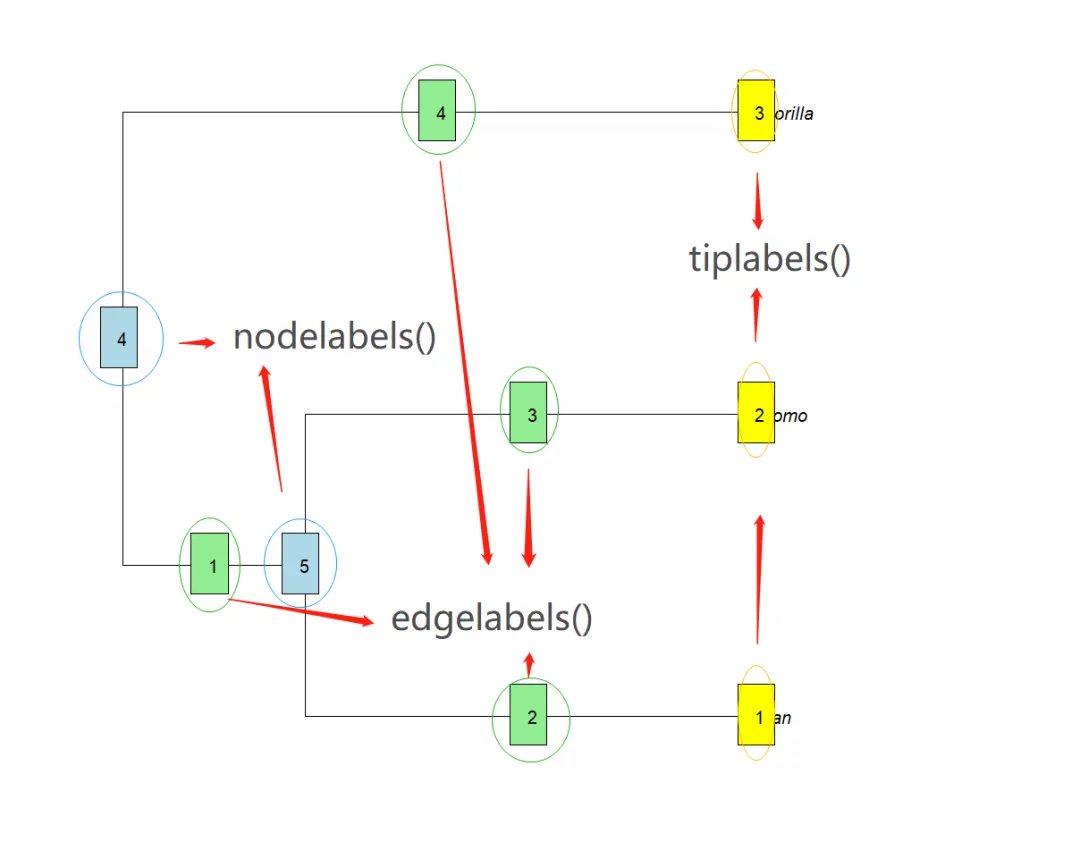
注意:上图的尖端(Tip)和节点(Node)处在同1个编号系统,即本质上都是Node:1. 处在”端”或”外侧”的Node是Tip,其它的是内部Node;2. 先编号Tip (顺序是从树图的下方至上方),后编号内部Node (从树图的上方至下方)。
这些函数提供了很多可能性:帮助页面中给出了一系列示例,这些示例可用example(nodelabels)来显示,此处不再赘述。这些函数(特别是nodelabels)的一个常见应用是绘制祖先值的饼图(Pie charts of ancestral values)。ape (例如ace)或其它软件包中的一些函数返回它们的祖先重建 (Ancestral reconstructions),这些重建沿着树的节点建立索引,因此使用nodelabels通常很简单直接。
与plot.phylo类似,这些选项可用于显示某些数据,或格式化注释 (Formatting the annotations)。选项text的工作方式与同名的标准函数相同;字符串的格式化使用col及其它选项完成(例如,font或cex可通过“…”传递)。除非frame = “none”,否则会在字符串周围绘制1个框。选项pch用于绘制标准绘图符号(Plotting symbols),取值范围为1~25,如下所示:
plot(rep(1:5, 5), rep(1:5, each = 5), pch = 1:25, xlim = c(1, 5.2),col = "blue", bg = "yellow", cex = 2)text(rep(1:5, 5) + 0.2, rep(1:5, each = 5), 1:25)
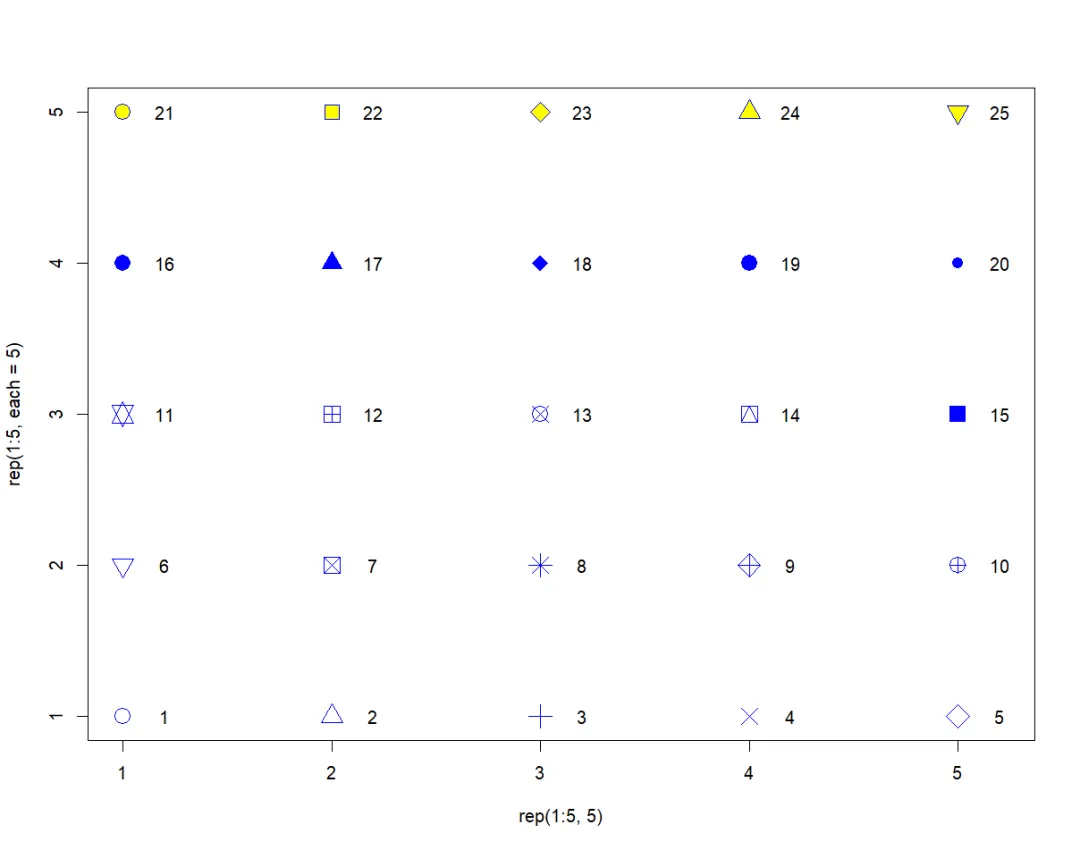
pch选项
应记住,这3个函数 (nodelabels、tiplabels、edgelabels?)都是低级制图命令,因此用户可使用可能的不同格式化选项多次调用它们,可能比使用单个调用更简单(可参考?nodelabels中的第1个例子)。
选项adj用于设置传递给text (以及用pch绘制的制图符号,与函数points不同) 的字符串的调整。这可用于打印节点周围的若干系列数字 (见?nodelabels)。根据每个字符串的大小进行调整,因此有用的值为:0(左对齐或下对齐)、0.5(居中)和1(右对齐或上对齐):
v <- c(0, 0.5, 1)layout(matrix(1:9, 3, 3))par(mar = c(3, 3, 3, 0), las = 1)for (i in v) {for (j in v) {plot(0, 0, "n", main = paste0("adj = c(", i, ", ", j, ")"))abline(v = 0, h = 0, lty = 3)text(0, 0, "Gorilla", adj = c(i, j))}}
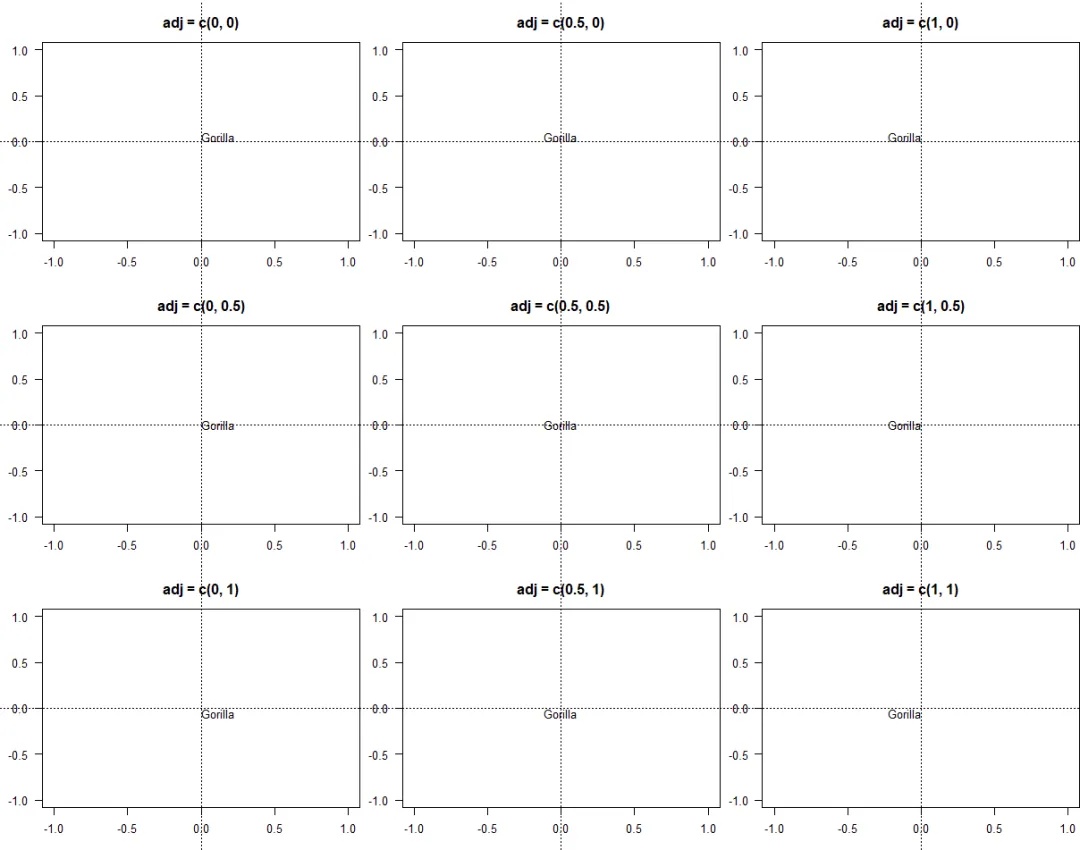
adj选项
也可接受其它值,但如果它们具有不同的(打印)宽度,则可能会导致字符串对齐不佳:
plot(rep(0, 3), 0:2, "n", las = 1)abline(v = 0, h = 0:2, lty = 3)text(0, 0:2, mytr$tip.label, adj = -1)text(0, 0:2, mytr$tip.label, adj = 2, font = 2)
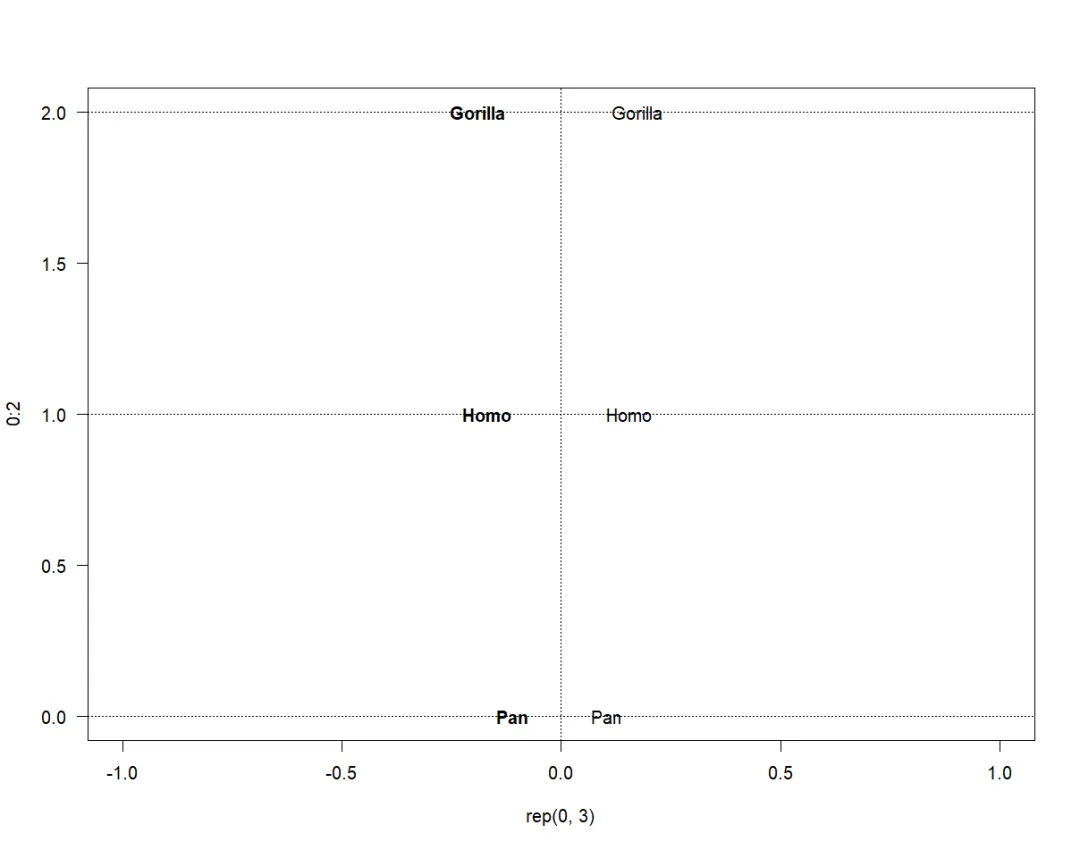
与前面的选项不同,选项pie和thermo读取矩阵,其中的值作为比例,以piecharts或thermometres显示。pies的“切片/slices”的颜色或thermometers的“水平/levels”用piecol指定(默认情况下,rainbow函数用于定义这些颜色)。选项horiz、width和height用于设置thermometres外观的格式。
4.3 在树旁对数据制图
下面2个函数在绘制好的树的旁边或连接处,添加1个图形 (如果树是环形/Circular的,则在其周围添加):
args(phydataplot)#function (x, phy, style = "bars", offset = 1, scaling = 1, continuous = FALSE,# width = NULL, legend = "below", funcol = rainbow, ...)# NULLargs(ring)#function (x, phy, style = "ring", offset = 1, ...)# NULL
它们的使用比以前的函数要复杂一些,因为在使用x.lim或y.lim绘制树时,通常需要留出一些空间(见前文)。要找到必须保留的空间可能并不简单:实际上,最简单的解决方案是尝试不同的值,可能使用如上所示的axis,或者使用plot.phylo的(不可见的)返回值。一旦为给定的数据集找到这些限制(区)的某些值,它们将在不同的图形设备(pdf(), png(), . . . )上以类似的方式工作。对于更复杂精细或自动化的解决方案,可以预先计算这些限制(见下文)。
这2个函数使用数据的标签(如果数据在向量中,则由它们的名称/names给出;如果数据在矩阵中,则由它们的行名称/rownames给出;如果数据在数据框中,则由它们的row.names给出)与树的尖端/Tip标签匹配/Match(参数phy)。因此,如果数据具有这样的(行)名称,则无需对其重新排序。
选项style可以采用7个不同的值,提供了很多可能性。此外,可以给出进一步的参数,这些参数随后被传递给graphics包的函数,从而提供定制化图形外观的灵活性。下表给出了style值,与phydataplot调用的graphics的函数,二者之间的对应关系:fancyarrows()在ape中:其参数与arrows()相同,加上选项type可以是“triangle”(默认)或“harpoon”(鱼叉状)

这里给出了一个灵长类树的简单示例,以说明使用type = “mosaic”可以做什么。我们构建一个包含10列的矩阵,其中填充值为1~6,并为其行和列命名:
x <- matrix(1:6, 3, 10)x# [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]# [1,] 1 4 1 4 1 4 1 4 1 4# [2,] 2 5 2 5 2 5 2 5 2 5# [3,] 3 6 3 6 3 6 3 6 3 6LETTERS[1:ncol(x)]# [1] "A""B""C""D""E""F""G""H""I""J"dimnames(x) <- list(c("Homo", "Gorilla", "Pan"), LETTERS[1:ncol(x)])
注意,这些数据的顺序与树的尖端标签的顺序不同:
x# A B C D E F G H I J# Homo 1 4 1 4 1 4 1 4 1 4# Gorilla 2 5 2 5 2 5 2 5 2 5# Pan 3 6 3 6 3 6 3 6 3 6mytr$tip.label# [1] "Pan""Homo""Gorilla"
可以将这些值视为类别/Categories或连续变量。对于phydataplot,这由选项continuous控制,continuous是1个逻辑值(默认为FALSE,因此变量被视为分类变量),或者是1个指定用于表示数据的类的数(The number of classes)的整数(如果continuous = TRUE,则使用10个类)。因为我们两次调用phydataplot,所以我们使用offset选项设置树和图形之间的空隙:
par(mar = c(10, 2, 5, 5))plot(mytr, x.lim = 30)phydataplot(x, mytr, "m", border = "white", offset = 2, width = 1)phydataplot(x, mytr, "m", border = "white", offset = 15,width = 1, continuous = 2)
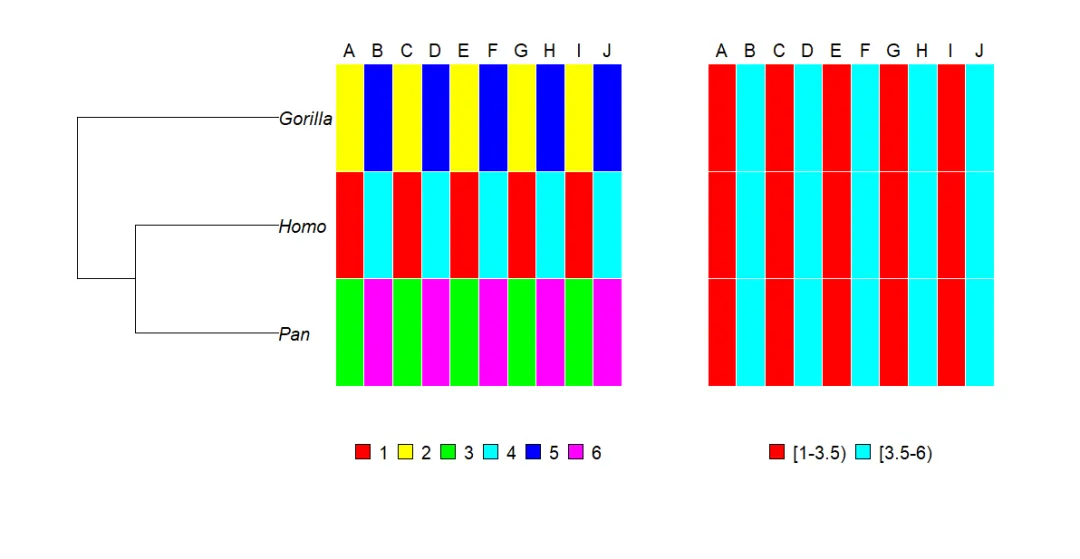
选项border = “white”和width = 1有助于获得更好看的图形。第1个”马赛克”(Mosaic)使用的配色方案便于检查矩阵的行和树的尖端之间的数据是否正确匹配。
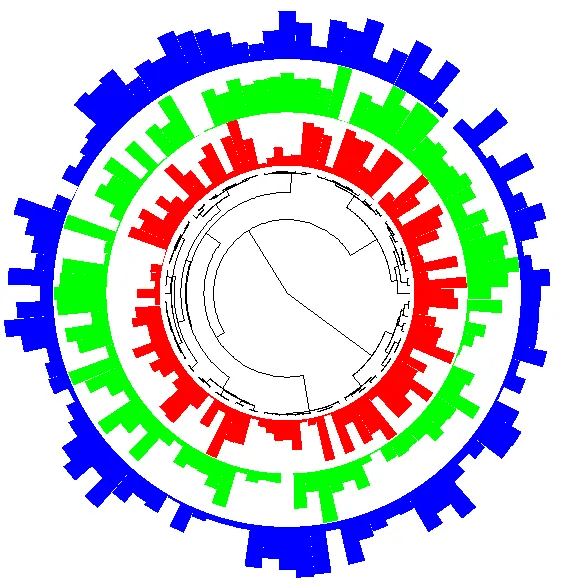
对于环形树,offset/偏移量是组合不同数据的“环”(rings)的关键,如下一个示例中的随机溯祖树(Random coalescent tree),该树具有100个尖端和均匀分布的3个变量:
n <- 100p <- 3set.seed(3)tr <- rcoal(n) # 随机溯祖树(Random coalescent tree)x <- matrix(runif(p * n), n, p)rownames(x) <- tr$tip.labelx# [,1] [,2] [,3]# t87 0.753599951 0.437275901 0.385993783# t93 0.984353975 0.764994170 0.553472379# t63 0.280397004 0.993582941 0.244482948# t37 0.862286026 0.754478029 0.117988848# t14 0.246813048 0.857998249 0.431723503# ... ...COL <- c("red", "green", "blue")par(xpd = TRUE)plot(tr, "f", x.lim = c(-5, 5), show.tip.label = FALSE, no.margin = TRUE)for (j in 1:p) ring(x[, j], tr, offset = j - 1 + 0.1, col = COL[j])
(附) ape中“phylo”类对象的结构


扫码、联系客服老师报名,领取资料、上手分析