 夜雨聆风
夜雨聆风我们的教程主要是以一个具体的例子作为线索,通过对公共数据库数据bulk-RNA-seq的挖掘,利用生物信息学分析来探索目标基因集作为某种疾病数据预后基因的潜能及其潜在分子机制,同时在单细胞水平分析(对scRNA-seq进行挖掘)预后基因的表达,了解细胞之间的通讯网络,以期为该疾病临床治疗提供新的参考,同时我们还可以经过分子对接实现药物靶点的初步筛选。
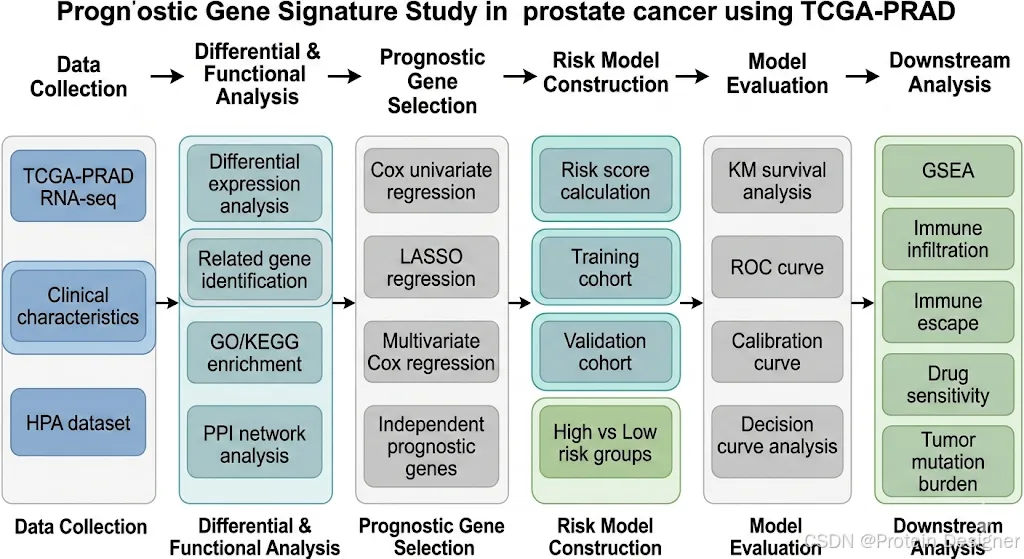
1.TCGA数据库
TCGA(The cancer genome atlas,癌症基因组图谱)由 National Cancer Institute(NCI,美国国家癌症研究所) 和 National Human Genome Research Institute(NHGRI,美国国家人类基因组研究所)于 2006 年联合启动的项目, 收录了各种人类癌症(包括亚型在内的肿瘤)的临床数据,基因组变异,mRNA表达,miRNA表达,甲基化等数据,是癌症研究者很重要的数据来源。
TCGA存储的数据包括SNV、转录组分析、生物样本信息、原始测序数据、CNV、DNA甲基化、临床信息等。这些数据可分为三个级别:1. 原始的测序数据(fasta,fastq等);2. 比对好的bam文件;3. 为经过处理及标准化的数据。
在具体数据下载时,可以直接从TCGA官网下载。
TCGA官网:https://portal.gdc.cancer.gov/
但是,由于TCGA数据库中的数据量过大,因此从TCGA官网中下载得到的数据经常是需要经过预处理的。虽然数据的获取较为容易,但是预处理比较困难。
因此,我们使用XENA数据库进行数据的获取。
Xena是一个在线网站,由美国加利福尼亚大学开发,主要用于探索TCGA(The Cancer Genome Atlas)和其他公共数据库的数据。Xena提供了一个直观的平台,使用户能够快速下载特定基因在特定癌症中的表达量数据,并进行可视化分析。
Xena官网:https://xenabrowser.net/datapages/
在最右边的窗口中,我们仅选择GDC Hub。
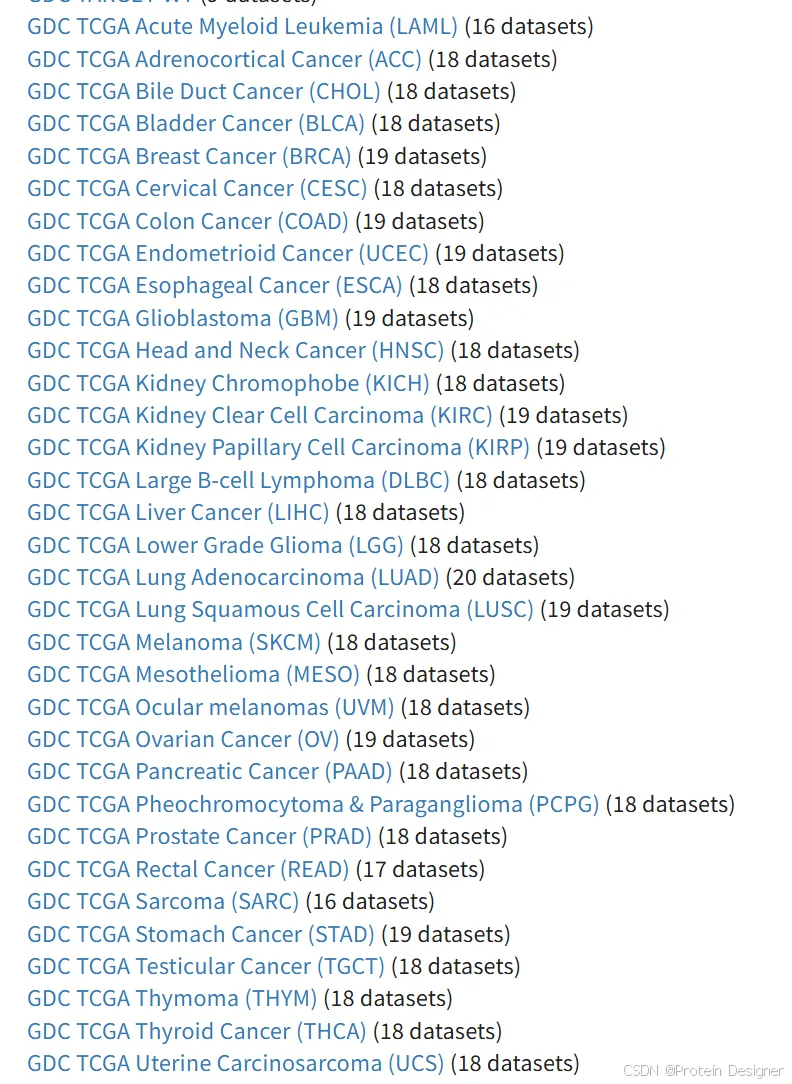
左侧的窗口中显示33种癌症的数据,需要选择课题对应的数据进行分析。
外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传
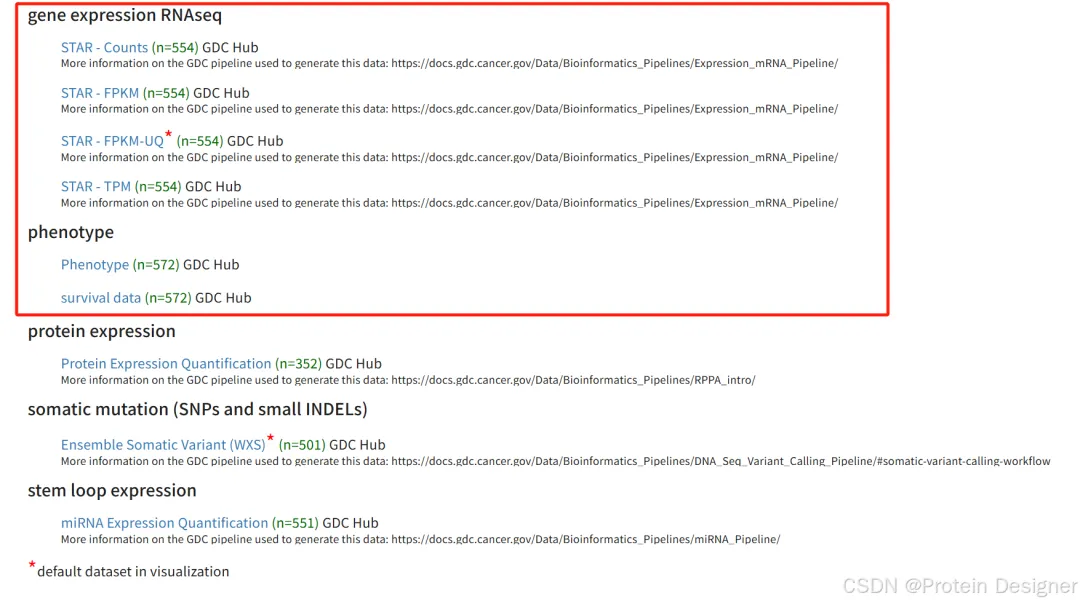
我们这里选择PRAD(前列腺癌)作为一个示例进行展示。如下图方框中展示的,我们需要Counts,FPKM,Phenotype,Survival这四个数据,点击下载即可。
2.数据下载及预处理
我们需要分别下载Counts(用于差异表达基因的计算)和FPKM(用于后续分析)数据。这里由于在服务器上使用Rstudio Server,因此直接使用Sestem命令去进行下载,也可以在浏览器中直接下载后,保存在当前文件夹。
# meta:https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-PRAD.clinical.tsv.gz
# data:https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-PRAD.star_fpkm.tsv.gz
system("wget -c https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-PRAD.clinical.tsv.gz")
system("wget -c https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-PRAD.star_counts.tsv.gz")
system("wget -c https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-PRAD.star_fpkm.tsv.gz")
system("wget -c https://gdc-hub.s3.us-east-1.amazonaws.com/download/gencode.v36.annotation.gtf.gene.probemap")
system("wget -c https://tcga-xena-hub.s3.us-east-1.amazonaws.com/download/survival%2FPRAD_survival.txt")
system("wget -c http://bioinfo.jialab-ucr.org/PCaDB/downloads/TCGA-PRAD_eSet.RDS")
同时由于大多数前列腺癌患者会接受治疗,大约三分之一的病例进展为晚期转移性前列腺癌,即进入了疾病的生命极限阶段。虽然近年来PCa的总体发病率有所下降,但是在过去十年中,诊断时的转移性疾病有所增加(此处为疾病原因,所以选择生化复发期(Biochemical Recurrence, BCR)作为预后的指标,这里需要根据具体情况去选择总生存期(Overall Survival,OS)和BCR。
OS(总生存期):指从治疗开始(如手术、放疗)或诊断之日起,到患者因任何原因死亡的时间。重点:反映患者的最终生存结局,是评估治疗效果的金标准。
BCR(生化复发):指治疗后(如根治性前列腺切除术或放疗)PSA水平再次升高,但尚未出现临床症状或影像学复发。重点:是早期复发指标,提示癌症可能进展,但不一定直接影响生存。
指标 BCR(生化复发) OS(总生存期) 定义 PSA升高(如术后PSA ≥0.2 ng/mL) 从治疗到死亡的时间 意义 早期复发信号,可能预示疾病进展 长期生存结果 影响因素 PSA监测灵敏度高,但并非所有BCR都会影响生存 受癌症进展、合并症、其他死亡原因影响 临床应用 用于早期干预(如挽救性放疗、激素治疗) 评估治疗的最终获益(如新药能否延长生命)
提取survival文件中的BCR信息及时间信息,并将他们加入到meta文件中。
library(limma)
library(dplyr)
# BCR
meta<-read.table("TCGA-PRAD.clinical.tsv.gz",header=T,sep="\t")
dat<-readRDS("TCGA-PRAD_eSet.RDS")
surv<-dat@phenoData@data
meta$sample1<-substr(meta$sample,1,15)
# meta[duplicated(meta$sample1),]$sample
# [1] "TCGA-HC-8265-01B" "TCGA-HC-8258-01A" "TCGA-HC-8261-01A" "TCGA-HC-7740-01B"
meta<-meta[!(meta$sample%in%c("TCGA-HC-8265-01B","TCGA-HC-8258-01B","TCGA-HC-8261-01B","TCGA-HC-7740-01B")),]
rownames(meta) <-meta$sample1
meta<-meta[surv$sample_id,]
rownames(surv) <-surv$sample_id
surv<-surv[meta$sample1,]
meta<-cbind(meta,surv[,c("time_to_bcr","bcr_status")])
colnames(meta)[c(90,91)] <-c("BCR.time","BCR")
meta$BCR.time<-meta$BCR.time*30
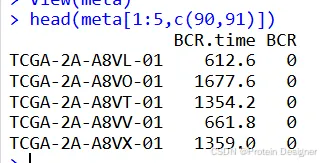
如下图,我们已经提取到了生存信息。
之后我们分别对FPKM和Counts数据进行处理。删除Not Reported的样本,使用gencode.v36.annotation.gtf.gene.probemap文件对基因名进行注释,之后保存成expr和meta文件。
data<-read.table("TCGA-PRAD.star_fpkm.tsv.gz",header=T,sep="\t")
data<-data.frame(row.names=data$Ensembl_ID,data[,c(2:ncol(data))])
colnames(data) <-stringr::str_replace_all(colnames(data),"\\.","-")
common.sample<-intersect(colnames(data),meta$sample)
data<-data[,common.sample]
meta<-meta[meta$sample%in%common.sample,]
table(meta$sample_type.samples,meta$vital_status.demographic)
# Alive Dead Not Reported
# Primary Tumor 483 10 2
# Solid Tissue Normal 52 0 0
meta<-meta[meta$sample_type.samples!="Metastatic"&meta$vital_status.demographic!="Not Reported",]
data<-data[,meta$sample]
gencode<-read.table("gencode.v36.annotation.gtf.gene.probemap",header=T)
rownames(gencode) <-gencode$id
colnames(gencode)[1] <-"ENSEMBL"
colnames(gencode) <-c("ENSEMBL","symbol")
gencode<-gencode[,-c(3,4,5,6)]
library(tibble)
data<-data%>%
as.data.frame() %>%
rownames_to_column("ENSEMBL") %>%
inner_join(gencode,"ENSEMBL") %>%
dplyr::select(-ENSEMBL) %>%#ENSEMBL一列多余删除
dplyr::select(symbol, everything()) %>%#gene列排在前排序
mutate(rowMean=rowMeans(.[grep('TCGA',names(.))])) %>%
arrange(desc(rowMean)) %>%#按照平均值从大到小排序
distinct(symbol,.keep_all=T) %>%#留下第一个gene
dplyr::select(-rowMean) %>%#反向选择去除rowMean这一列
tibble::column_to_rownames(colnames(.)[1]) #把第一列gene变成行名并删除
write.csv(data,"train_Data_PRAD.expr.csv",row.names=T,quote=T)
write.csv(meta,"train_Data_PRAD.meta.csv",row.names=T)
# PRAD-counts
meta<-read.table("TCGA-PRAD.clinical.tsv.gz",header=T,sep="\t")
dat<-readRDS("TCGA-PRAD_eSet.RDS")
surv<-dat@phenoData@data
meta$sample1<-substr(meta$sample,1,15)
# meta[duplicated(meta$sample1),]$sample
# [1] "TCGA-HC-8265-01B" "TCGA-HC-8258-01A" "TCGA-HC-8261-01A" "TCGA-HC-7740-01B"
meta<-meta[!(meta$sample%in%c("TCGA-HC-8265-01B","TCGA-HC-8258-01B","TCGA-HC-8261-01B","TCGA-HC-7740-01B")),]
rownames(meta) <-meta$sample1
meta<-meta[surv$sample_id,]
rownames(surv) <-surv$sample_id
surv<-surv[meta$sample1,]
meta<-cbind(meta,surv[,c("time_to_bcr","bcr_status")])
colnames(meta)[c(90,91)] <-c("BCR.time","BCR")
meta$BCR.time<-meta$BCR.time*30
data<-read.table("TCGA-PRAD.star_counts.tsv.gz",header=T,sep="\t")
data[,2:ncol(data)] <-2^as.matrix(data[,2:ncol(data)])-1
data<-data.frame(row.names=data$Ensembl_ID,data[,c(2:ncol(data))])
colnames(data) <-stringr::str_replace_all(colnames(data),"\\.","-")
common.sample<-intersect(colnames(data),meta$sample)
data<-data[,common.sample]
meta<-meta[meta$sample%in%common.sample,]
table(meta$sample_type.samples,meta$vital_status.demographic)
# Alive Dead Not Reported
# Primary Tumor 483 10 2
# Solid Tissue Normal 52 0 0
meta<-meta[meta$sample_type.samples!="Metastatic"&meta$vital_status.demographic!="Not Reported",]
data<-data[,meta$sample]
gencode<-read.table("gencode.v36.annotation.gtf.gene.probemap",header=T)
rownames(gencode) <-gencode$id
colnames(gencode)[1] <-"ENSEMBL"
colnames(gencode) <-c("ENSEMBL","symbol")
gencode<-gencode[,-c(3,4,5,6)]
data<-data%>%
as.data.frame() %>%
rownames_to_column("ENSEMBL") %>%
inner_join(gencode,"ENSEMBL") %>%
dplyr::select(-ENSEMBL) %>%#ENSEMBL一列多余删除
dplyr::select(symbol, everything()) %>%#gene列排在前排序
mutate(rowMean=rowMeans(.[grep('TCGA',names(.))])) %>%
arrange(desc(rowMean)) %>%#按照平均值从大到小排序
distinct(symbol,.keep_all=T) %>%#留下第一个gene
dplyr::select(-rowMean) %>%#反向选择去除rowMean这一列
tibble::column_to_rownames(colnames(.)[1]) #把第一列gene变成行名并删除
rowname<-rownames(data)
data<-apply(data, 2, as.integer)
rownames(data) <-rowname
write.csv(data,"train_Data_PRAD.expr.counts.csv",row.names=T,quote=T)
write.csv(meta,"train_Data_PRAD.counts.meta.csv",row.names=T)
最终,我们下载并预处理得到的文件格式如下:
> meta[1:5,1:4]
sample id disease_type case_id
TCGA-2A-A8VL-01 TCGA-2A-A8VL-01A 49197847-cc83-4ce1-8397-d09cea4c4928 Adenomas and Adenocarcinomas 49197847-cc83-4ce1-8397-d09cea4c4928
TCGA-2A-A8VO-01 TCGA-2A-A8VO-01A 91c0d161-2b59-4b7a-8c19-6d26dea31849 Adenomas and Adenocarcinomas 91c0d161-2b59-4b7a-8c19-6d26dea31849
TCGA-2A-A8VT-01 TCGA-2A-A8VT-01A 931b549f-b9f2-4e8d-83ed-ff663671883c Adenomas and Adenocarcinomas 931b549f-b9f2-4e8d-83ed-ff663671883c
TCGA-2A-A8VV-01 TCGA-2A-A8VV-01A 75a7afb5-66d5-47e3-8a8a-3e3a1e749a96 Adenomas and Adenocarcinomas 75a7afb5-66d5-47e3-8a8a-3e3a1e749a96
TCGA-2A-A8VX-01 TCGA-2A-A8VX-01A 942f1788-d977-4ac0-a177-659f9d4cd077 Adenomas and Adenocarcinomas 942f1788-d977-4ac0-a177-659f9d4cd077
> data[1:5,1:5]
TCGA-2A-A8VL-01A TCGA-2A-A8VO-01A TCGA-2A-A8VT-01A TCGA-2A-A8VV-01A TCGA-2A-A8VX-01A
MT-CO3 13.04807 14.06800 11.91562 12.91113 13.72980
MT-CO2 12.98903 14.36177 11.99809 12.96906 13.96033
MT-ND4 13.14180 14.04815 12.09947 12.76331 14.04760
MT-CO1 13.25132 13.16147 12.06882 12.59348 12.83341
MT-ND3 12.66159 13.81111 11.44092 12.34254 13.92339
得到的样本数统计:495例前列腺癌肿瘤组织样本和52例癌旁对照组织样本。差异分析基于所有疾病样本和对照样本做, 预后模型构建及基于高低风险组或风险评分相关分析基于有生化复发时间的样本(461例)做。
