 夜雨聆风
夜雨聆风今天在对一个芯片数据做差异分析、绘火山图时,直接踩坑了 —— 出来的图完全没法看,太离谱了!X 轴范围直接飙到 -2e+05 ~ +1e+05,也就是从-20 万到+10 万;原本该分开的Up、Down 基因,全被挤压在 X 轴 0 点附近,所有点挤成一条竖线,压根分不清谁是上调、谁是下调,更别提看真实的表达倍数变化了。明明左侧标注的是下调基因(比如 MT1L、APOC3),右侧是上调基因(比如 MIF、SPINK1),但因为 X 轴异常,完全体现不出它们的表达差异,等于白做了一遍分析。
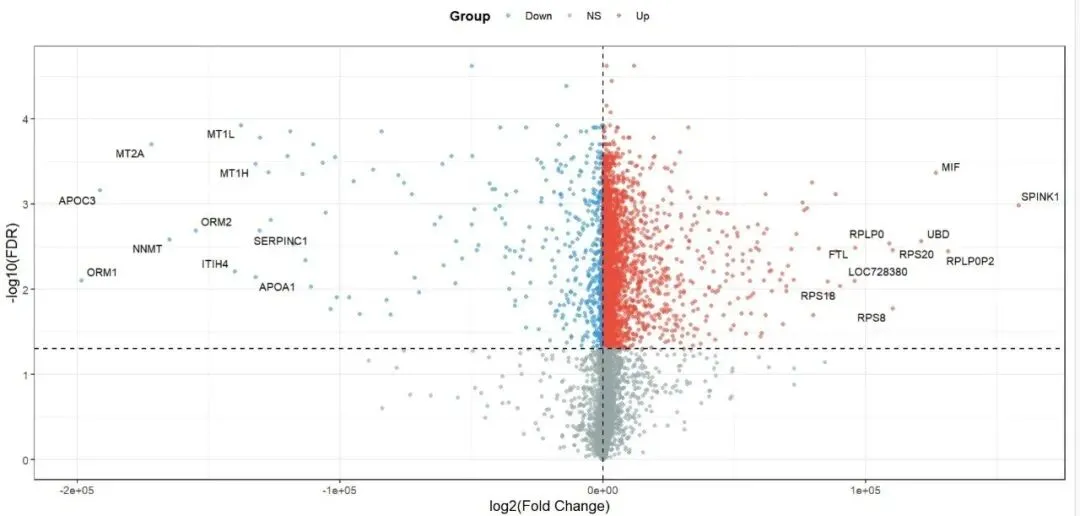
异常的火山图
盯着这张异常的火山图看了半天,查了一堆资料,才发现问题出在差异分析前的表达矩阵预处理 —— 我没有给数据做 log2 转换!要知道,GEO 芯片数据必须先做 log2 标准化,才能用 limma 包跑差异分析,我直接用未标准化的矩阵跑了 limma,导致 logFC 计算完全出错,后续画图用的都是错误的 logFC,最后才出了这么个 “四不像” 的图。
这时候我就疑惑了:我明明是从 GEO 官网下载的表达矩阵文件(就是那个 GSE*_series_matrix.txt.gz 格式),按说应该是处理好的,怎么会出现这种情况?后来才搞明白,GEO 下载的这个文件,90% 的情况确实已经做了 log2 标准化,直接用就行,但有一部分数据集是原始信号,没做 log2 转换,必须自己手动处理。这里插个小提醒:如果从 GEO 下载的是 CEL 格式的原始芯片文件,那肯定是未标准化的,必须用 rma () 函数做背景校正+标准化+log2 转换,一步都不能少。
解决完眼前的问题,我又琢磨:以后再遇到这种情况,怎么快速判断读入的表达矩阵是不是已经标准化了?结合自己踩的坑,整理了最实用的方法(亲测),分享给大家。
一、先搞懂核心原理(简单好记,不用死记硬背)
1.原始未标准化矩阵(不管是芯片还是测序原始数据)
数值范围特别大,从 0 到几万、甚至几十万都有可能(比如芯片的原始信号、测序的原始 counts),特点就是数值跨度极大,不同基因的表达量差异悬殊,一眼就能看出来不对劲。
2. 标准化后矩阵(可以直接跑差异分析的)
芯片数据:经过 log2 转换后,数值范围通常在 4-16 之间(这是 Affymetrix 芯片的标准范围);测序数据:用 vst 或 rlog 转换后,范围大概在 -5-15 之间;特点是数值集中、分布均匀,基本符合正态分布,看起来很规整。
二、最快最准的判读方法:3 行代码搞定
#直接运行这3行代码min(expr_matrix, na.rm = T) # 最小值max(expr_matrix, na.rm = T) # 最大值median(expr_matrix, na.rm = T) # 中位数
针对芯片数据,判断标准很简单:
最大值 > 100,中位数 < 10 → 原始未标准化 ❌ 必须做 log2 转换
最大值 5-20,中位数 8-12 → 已做 log2 标准化 ✅ 可直接跑差异分析
三、如果判断是未标准化,怎么补做?(直接抄代码)
# 补做log2转换(+1避免log(0)报错)expr_matrix_log2 <- log2(expr_matrix + 1)
2.测序数据(RNA-seq counts):
# 用DESeq2做vst标准化(推荐)library(DESeq2)dds <- DESeqDataSetFromMatrix(countData = expr_matrix,colData = sample_info,design = ~ group)dds <- DESeq(dds)expr_matrix_vst <- vst(dds, blind = FALSE)
补做完成后,再用前面的 3 行代码检查一下数值范围,确认标准化到位了,再跑差异分析、绘火山图就不会出错了。
最后补充:如何一眼判断火山图是否正常?
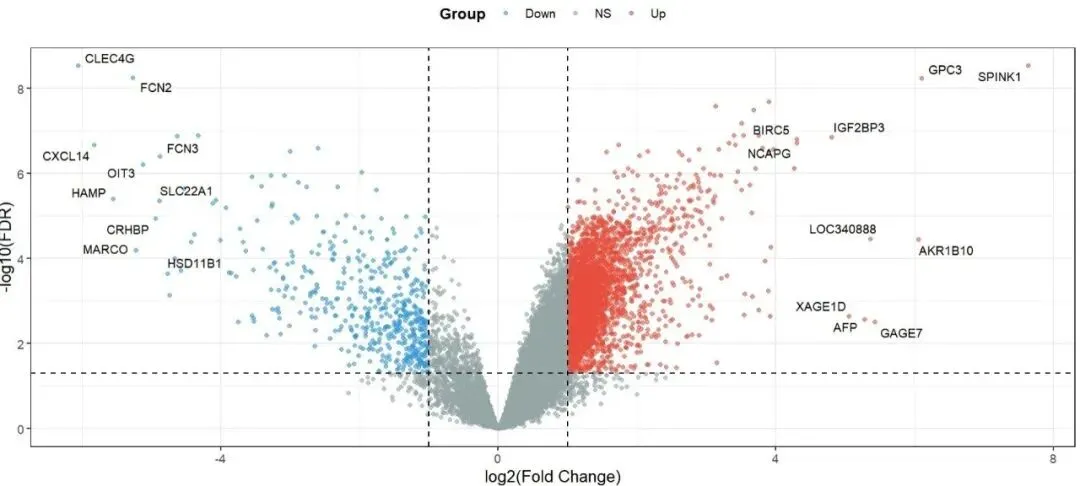
✅ 正常的标准火山图,应该是这样的:大部分基因集中在中间(灰色,无显著差异),显著上调的基因(红色)在右侧,显著下调的基因(蓝色)在左侧,整体呈 “火山喷发” 的形状,越往上的点,差异越显著。
❌ 像我这次遇到的异常形态:X 轴范围巨大(± 几万、几十万),所有基因挤成一条竖线,完全没有火山形状,九成以上是因为没做 log2 标准化,或者标准化操作出错了。
好在最后修正了标准化步骤,重新跑了分析,画出了正常的火山图,也算踩坑踩出了经验。
本文仅为个人科研学习经验分享与踩坑记录,仅供学习交流使用,不构成任何科研指导建议。不同数据集情况存在差异,实操时请结合自身数据合理判断、谨慎使用。
#GEO 芯片分析 #log2 标准化 #火山图绘制#科研踩坑#表达矩阵预处理#R 语言代码 #差异分析#研究生科研
【往期精彩】
