 夜雨聆风
夜雨聆风上期回顾
药品技术转移 SOP 模板 + 全流程实操要点(三方权责厘清版)
引言
药品技术转移完成工艺制备、检验方法全流程技术转移后,稳定性研究是佐证新旧场地产品质量等同性、保障上市药品质量稳定的核心环节,也是药品场地变更、委托生产变更、生产线调整过程中,注册申报与现场核查重点核查内容。
技术转移绝非简单工艺移交,唯有同步完成稳定性数据对标核验,才能证实转移后产品贮藏特性、有效期、质量变化趋势与原生产场地保持一致,从源头规避质量风险,满足合规管理要求。
原料药物与制剂、生物制品的稳定性试验目的,见图1
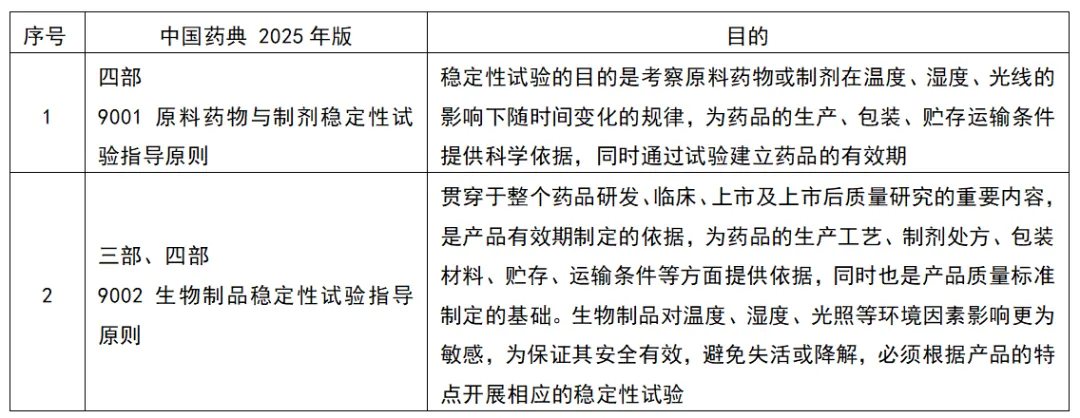
图1 原料药物与制剂、生物制品的稳定性试验目的
一、稳定性试验参考依据
1、《中国药典》2025 年版:9402 生物制品稳定性试验指导原则;9001 原料药物与制剂稳定性试验指导原则
2、ICH Q1A (R2)《新原料药和制剂的稳定性试验》
3、《药品生产质量管理规范(GMP)》2010 年版“第十章质量控制与质量保证”中“第三节持续稳定性考察”
4、企业内部SOP:《药品稳定性考察管理规程》《偏差管理规程》《OOS/OOT 处理规程》《数据完整性管理规程》
二、技术转移阶段开展稳定性研究核心目的
1、验证接收场地量产工艺生产的产品,与转出场地产品质量属性、降解趋势保持一致
2、确认现有内包材、外包装体系,在新生产环境、仓储环境下防护性能稳定可靠
3、复核产品既定贮藏条件、有效期时长无需调整,保证市场流通药品有效期合规
4、排查场地环境、生产设备、操作人员、生产批量变更带来的隐性质量隐患
5、形成完整可追溯的稳定性研究资料,应对药品核查、注册变更、日常质量审计
6、建立接收企业常态化稳定性考察体系,完善全生命周期药品质量管控
三、需开展稳定性比对研究的常见转移场景
1、药品整体生产场地搬迁、异地新建生产线完成技术移交
2、MAH 持有人变更受托生产企业,完成全品种技术转移
3、核心生产设备更新换代、生产批量发生重大调整
4、处方辅料微调、关键生产工艺优化落地后正式转产
5、产品内外包装材料更换同步完成技术转移
6、质量控制实验室异地迁移,检验体系完成移交确认
7、特殊气候区域投产,需适配地域温湿度开展稳定性考察
8、生物制品原液 / 制剂生产线转移,需同步开展工艺一致性稳定性验证
四、技术转移阶段各类稳定性试验实操侧重
1. 影响因素试验(强降解试验)
以极端环境条件考察产品固有稳定性,技术转移阶段优先复核开展,对比新旧场地样品高温、高湿、光照、冻融条件下质量变化,明确产品易降解项目、敏感影响因素,统一风险防控要点。
2. 加速稳定性试验
作为短期质量预判核心试验,采用商业化正式批次样品,统一放置环境与检测频次,快速比对杂质增长、含量变化、性状改变等数据,初步判定工艺转移后产品质量稳定性是否达标。生物制品需采用温和加速条件,避免非典型降解。
3. 长期稳定性试验
贴合市场实际贮藏条件开展,为药品有效期核定、日常留样考察提供核心数据,技术转移后同步启动长期留样,长期追踪质量变化趋势,与原场地历史数据横向对标。生物制品需严格按照 2-8℃/≤-20℃等实际贮藏条件开展。
4. 特殊条件试验
针对生物制品、注射剂等特殊剂型,需额外开展冻融稳定性、机械稳定性、包装相容性等专项试验,评估运输、灌装、使用过程中的质量风险。
五、技术转移稳定性试验统一规范要求
1、试验样品:选取正式商业化生产规模合格批次,处方、工艺、投料比例与原场地完全一致,生物制品需采用同工艺、同表达体系生产的原液 / 制剂
2、包装形式:严格采用上市销售统一包装,不得使用简易包装开展试验,生物制品需重点评估包材吸附、析出风险
3、环境条件:温湿度、光照、放置方式完全对标转出企业原有试验标准,生物制品需额外控制冻融次数、振荡条件
4、检测依据:必须使用已完成分析方法转移、确认合格的检验方法开展检测,生物活性、纯度等关键项目需完成方法验证
5、人员仪器:检验人员完成实操培训,检测仪器完成校准确认,生物试验操作人员需完成带教与盲样考核
6、记录管理:全程留存原始记录、检测图谱、温湿度监控数据,做到全程可溯源,生物试验需完整记录孵育条件、加样过程
六、稳定性试验通用核心考察项目
(一)化药 / 中药通用项目 见图2

图2化药 / 中药稳定性试验通用项目
(二)生物制品专属核心考察项目 见图3

图3 生物制品稳定性试验专属项目
七、新旧场地稳定性数据比对基本原则
1、质量降解整体趋势保持一致,无反向异常变化
2、有关物质 / 杂质增长速率处于合理等同区间,生物制品聚体、降解片段变化趋势需与原场地数据一致
3、主成分含量 / 生物活性下降幅度符合原有波动范围
4、所有考察项目变化趋势均在质量标准限度内,无超出内控预警线的情况
5、出现数据偏差及时启动调查分析,明确偏差原因并落实整改,生物试验数据波动需重点排查试验条件、人员操作差异
6、建立稳定性数据趋势分析台账,定期汇总研判,生物制品需额外建立活性、聚体等关键指标的趋势预警机制
八、生物制品稳定性研究特殊试验要求
1. 冻融稳定性考察
针对需低温 / 冷冻保存的原液或制剂,必须设计冻融试验,考察反复冻融对产品活性、聚体形成的影响,明确允许的冻融次数,为生产、运输、使用过程中的风险控制提供依据。
2. 机械稳定性考察
针对注射剂、预充针等剂型,需模拟运输、灌装过程中的振荡、剪切力,评估产品的聚集风险,避免因机械应力导致的质量下降。
3. 加速条件的特殊选择
生物制品通常采用更温和的加速条件(如 25℃±2℃/60% RH±5% RH),部分蛋白药物甚至需在低温下进行加速考察,避免高温导致的非典型降解,影响有效期评估的准确性。
4. 包装系统相容性考察
生物制品对包装材料的相容性要求更高,需评估包材吸附、析出、密封完整性对产品稳定性的影响,尤其是单剂量制剂、预充针等直接接触产品的包装。
九、技术转移稳定性研究常见合规隐患
(一)化药 / 中药通用隐患
1、仅开展短期加速试验,未同步启动长期稳定性对标考察
2、试验温湿度、放置条件与原场地不统一,数据失去比对参考意义
3、分析方法尚未完成转移确认,直接用于稳定性样品检测
4、稳定性留样数量不足、留样仓储环境不符合规范要求
5、未建立常态化趋势分析机制,异常质量变化未能提前预判
6、稳定性研究资料零散缺失,无法形成完整闭环备查资料
7、盲目沿用原有有效期,未结合转移后试验数据重新复核确认
(二)生物制品专属核查缺陷
1、仅考察理化指标,遗漏生物活性、聚体、电荷异构体等关键 CQA 项目
2、加速 / 长期试验条件与产品实际贮藏条件不符,如将 - 20℃保存的产品放在 25℃下做加速试验
3、冻融次数未做考察,或考察次数不足,无法支撑产品运输、使用过程中的风险评估
4、检测方法未完成转移,直接用接收方实验室数据替代转出方数据,无可比性
5、稳定性数据异常(如活性骤降、聚体快速增加)未启动调查和 CAPA,直接忽略
6、机械稳定性、包装相容性等专项试验缺失,无法评估运输风险
十、附录
药品稳定性试验研究方案(模板)及其4个附件
由于篇幅有限,稳定性研究方案(终版)、接收 / 转出场地工艺参数对比表、稳定性试验原始记录、试验设备校准确认记录 已经整理成word。可供参考。
如需要,领取方式:在公众号“李志红 LBC”,在主页 “发消息” 对话框回复关键词 “稳定性”,即可获取网盘链接与提取码。
#药品技术转移#药品稳定性研究#GMP质量管控#药企QA干货#药品合规管理
