 夜雨聆风
夜雨聆风上期回顾
药品技术转移 SOP 模板 + 全流程实操要点(三方权责厘清版)
上一篇我们分享了《药品技术转移SOP 模板 + 全流程实操要点》,已将立项、资料移交、工艺验证、偏差管控、三方权责等核心环节完整落地。
质量转移无小事,数据合规无侥幸。
方法转移拼的不是数据合格,而是风险闭环。
一地方法,两地验证,严控异常,守住质量底线。
在飞检与注册核查中,分析方法转移环节高频问题频发:方法未转移直接借鉴、验证项目不完整、数据缺乏可比性、OOS / 偏差 / OOT 处理不规范、报告资料不规范等,极易导致补验证、整改,甚至影响注册申报进度。
本文依据中国药典、CDE 及 ICH 指导原则,对分析方法转移进行全流程拆解,覆盖化学方法与微生物方法,配套可参考借鉴的模板,使用时需要结合产品的注册标准和检测依据。
一、分析方法转移概述
分析方法转移,是转出方(方法建立实验室)向接收方(方法使用实验室)传递检验方法的过程,通过同步试验与数据比对,证明接收方可稳定、准确、可重复执行方法,确保两地检测结果一致、可比对。
核心目的
保障技术转移前后质量控制标准统一
防范方法不可重现引发的 OOS、偏差、OOT与核查缺陷
满足 GMP、ICH Q2、中国药典、CDE 相关合规要求
适用场景
药品技术转移、生产场地变更(MAH 委托生产、异地扩产)
实验室搬迁、关键仪器更换、核心人员变动
注册工艺变更涉及检测方法调整
原辅料、中间体、成品质量标准转移
无菌、微生物限度、细菌内毒素等微生物检验方法转移 / 变更
二、权威参考依据:见图1
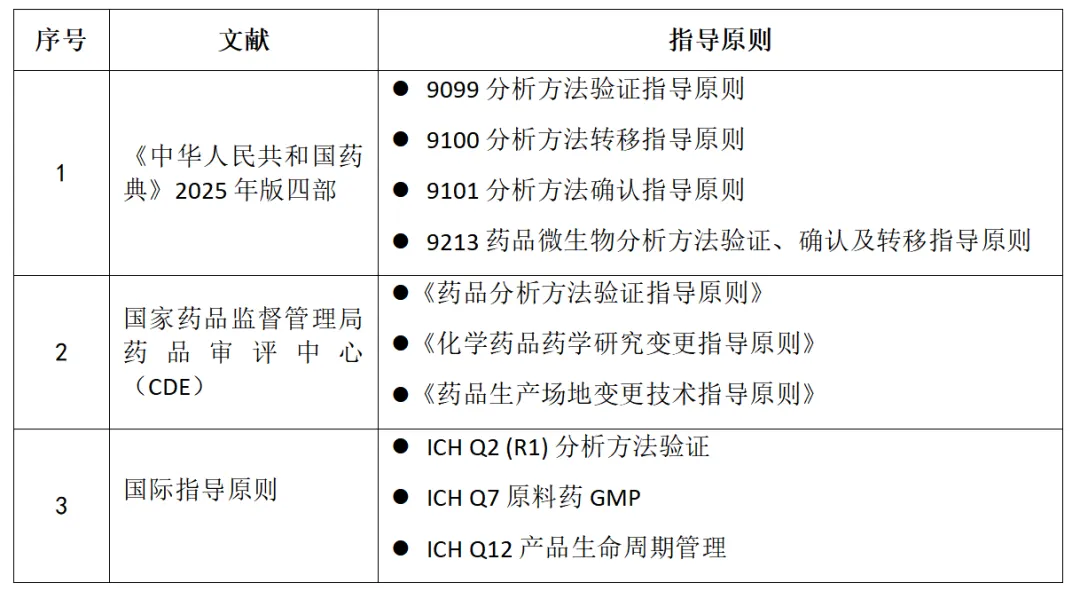
图1 分析方法转移参考依据
三、核查必考:3 种分析方法转移方式怎么选?见图2
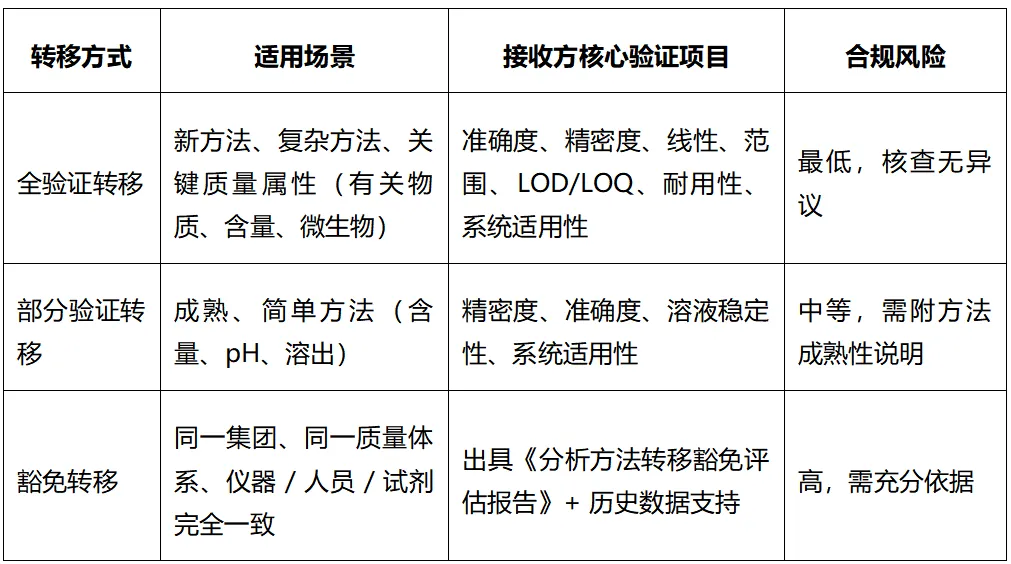
图2 3 种分析方法转移方式
四、分析方法转移全流程 5 步走(零踩坑)
第 1 步:转移前准备(资料移交 + 条件确认)
转出方提供:方法SOP、验证报告、色谱图 / 光谱图、标准品 / 对照品信息、试剂耗材规格
接收方确认:仪器型号、色谱柱、试剂批次、环境温湿度、人员资质
双方签署《资料移交清单》,明确责任边界
第 2 步:制定《分析方法转移方案》
明确转移项目、批次(≥2 批)、浓度水平(≥ 3 个)
规定可接受标准(如 RSD、回收率、偏差范围)
明确试验步骤、数据记录要求、数据比对方法
双方 QA 审核批准后再启动试验
第 3 步:同步试验与数据比对
转出方与接收方同步测试同一样品,避免批次差异影响
完整记录原始数据、图谱、系统适用性结果,全程可追溯
计算 RSD、回收率、两组数据偏差,判断是否符合标准
第 4 步:结果评估、OOS /OOT /偏差处理
结果符合标准与趋势要求:判定转移通过,进入报告归档阶段。
结果出现异常时,需区分以下两种情况启动调查:
OOS(超出标准结果):检验结果直接超出质量标准规定的限度(如含量、有关物质等指标不达标),必须立即启动OOS 调查,按企业 SOP 开展偏差处理,排查仪器、试剂、人员操作、方法适用性、样品均一性等原因,并制定纠正与预防措施(CAPA)。
第 5 步:出具《分析方法转移报告》与 GMP 归档
转出方与接收方同步测试同一样品,避免批次差异影响
报告包含:转移背景、试验方案、数据结果、评估结论、偏差变更记录
附件:所有原始图谱、数据记录、审批签字文件
双方QA 审核签字,按 GMP 要求归档,随时可迎检
五、接收方必做核心项目 + 通用可接受标准
必做项目(核查必看)
系统适用性试验
精密度(重复性、中间精密度)
准确度(回收率试验)
溶液稳定性(考察样品 / 标准品在分析过程中的稳定性)
转出方与接收方数据比对
通用可接受标准,见图3
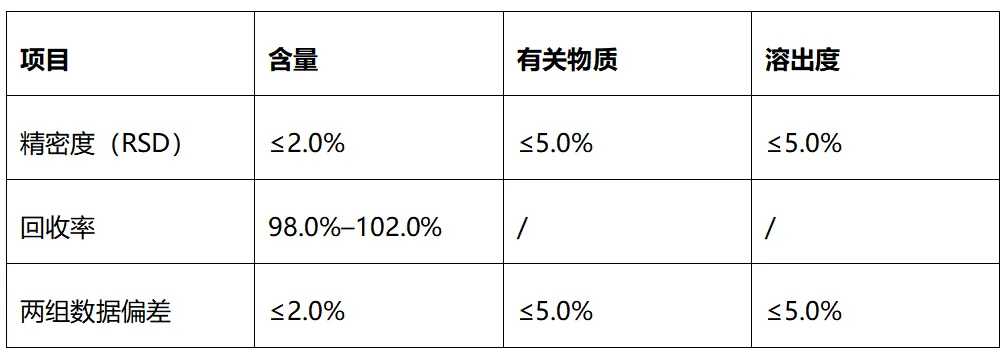
图3 分析方法接收方通用可接受标准
六、微生物分析方法转移,见图4
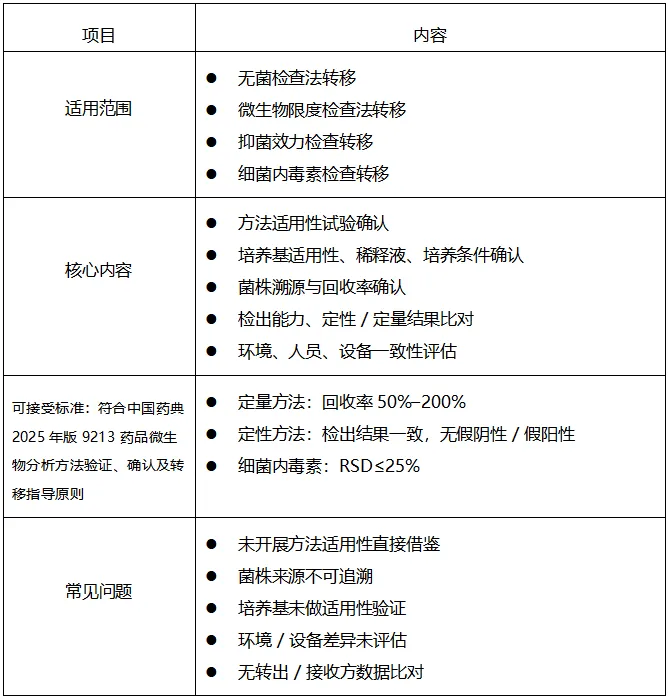
图4 微生物分析方法转移内容
七、生物制品分析方法转移专属要点
生物制品涵盖原液、制剂、细胞类产品、抗体、疫苗、重组蛋白等,其检验体系与化药差异极大,方法转移不能直接参照化学药品逻辑,需单独统筹确认。
(一)生物制品方法转移核心特点
1、检验多以生物活性、效价、细胞水平检测、免疫学方法为主,理化项目占比偏低
2、试验结果受细胞株状态、实验动物、血清批次、操作人员手法、孵育环境影响极大,波动性高于理化检测
3、方法耐受性偏弱,试验条件细微偏差即可导致数据偏差,转移确认要求更严苛
4、部分活性检测无绝对真值,侧重趋势一致、区间吻合、平行性稳定
(二)生物制品常见需转移核心项目
生物学活性/ 比活性测定
效价测定、体外体内效力试验
蛋白含量、纯度、分子量、等电点
宿主细胞蛋白残留、DNA 残留检测
抗体结合活性、结合滴度检测
无菌检查、热原、细菌内毒素
异常毒性、过敏试验等安全性检查
抗原含量、佐剂相关检测
(三)生物制品优先选用的转移方式
比对试验法(首选主流)
转出方与接收方同步同批样品平行测定,对比均值、RSD、区间范围、阳性阴性判定一致性,最贴合生物试验特性
全方法再验证
针对新建实验室、首次承接品类,接收方完整重做方法学验证,确认自身实验室条件可稳定承载检测
部分确认
仅用于成熟通用筛查方法,活性类项目不建议简化确认
再培训+ 实操考核
细胞培养、动物试验、酶联免疫等强实操项目,必须完成人员实操带教、盲样考核合格后方可落地
(四)生物制品方法转移关键控制要点
1、实验体系统一
细胞株代次、培养基批号、胎牛血清来源、孵育温湿度、孵育时长全部对齐转出方标准,严禁随意调整
2、标准品/ 参考品同源
统一使用同批次国家基准品、工作参考品,建立统一标定溯源体系
3、操作人员固定
化生物试验手感经验影响大,尽量固定专职检验人员,减少人员轮换带来的数据波动
4、试验环境分区管控
无菌区、细胞培养区、活性检测区环境级别、洁净度、温湿度保持一致
5、数据判定标准统一
临界结果、边缘数据、阴阳判定、结果修约规则提前统一口径,避免判定分歧
(五)生物制品转移常见易疏漏问题
1、只转移理化项目,遗漏生物活性、残留杂质核心项目确认
2、细胞传代习惯、接种密度、加样节奏未统一,导致活性数据差异偏大
3、内毒素、无菌等合规项目简化确认,后期核查存在隐患
4、未建立生物试验异常结果调查流程,出现波动无法溯源
5、未同步转移阳性对照、阴性对照制备与保存规范
八、飞检 / 注册核查高频缺陷清单(避坑重点)
(一)化药、中药
1、无分析方法转移方案/ 报告,仅凭口头交接
2、仅做含量转移,忽略有关物质、溶出、微生物等关键项目
3、无转出方与接收方的同步比对数据,仅凭接收方数据判定通过
4、仪器、色谱柱、试剂不一致,未做风险评估
5、OOS / 偏差 / OOT未调查、无 CAPA,直接忽略异常数据
6、方法转移报告不完整,缺图谱、缺原始数据、缺签字
7、豁免转移无依据、无评估报告,仅凭经验判断
8、接收方擅自修改方法参数
9、仅关注OOS(超出标准结果)的调查处理,未建立OOT(超出趋势结果)的监控、判定与调查机制,忽略结果趋势异常带来的隐性质量风险,不符合过程控制与持续改进的要求。
(二)生物制品专属高频缺陷(核查重点)
1、重理化、轻生物活性:方法转移仅确认理化项目,生物学活性、细胞法、免疫学检测未做比对试验,直接沿用转出方数据,属于严重缺陷。
2、生物试验条件未对齐:细胞代次、血清批号、孵育时间、加样手法未统一,导致活性数据波动大,无可比性。
3、标准品 / 参考品管理不规范:工作标准品未同步转移、未标定、无溯源链,残留类项目(DNA、HCP)判定依据不足。
4、人员能力无考核记录:生物检测操作人员无带教记录、无盲样比对考核,检查员判定人员不具备检测资质。
5、边界数据处置随意:生物试验临界值、边缘结果无统一判定规则,复测无依据、无偏差调查。
6、方法确认项目不全:宿主残留、内毒素、无菌等合规筛查项目简化确认,未满足药典三部及核查要求。
7、原始记录过于简单:细胞培养、酶联反应、孵育时长无实时记录,缺少过程追溯性
九、附录:6个参考模板(领取方式见文末或评论区)
附录 1:分析方法转移方案框架
附录 2:分析方法转移报告模板
附录 3:分析方法转移豁免评估表
附录 4:方法转移 OOS / 偏差处理流程
附录 5:微生物方法转移参考模板
附件6:生物制品比对确认表模版
总结
方法转移不在于“做合格数据”,而在于管住OOS/偏差/OOT全链条风险;合规不留空白,波动不漏隐患,才是方法转移的底层逻辑。
下一篇将拆解:技术转移中的稳定性研究要点,持续整理核查参考要点,助力合规迎检。
参考依据
《中华人民共和国药典》2025 年版三部、四部
国家药监局CDE 相关指导原则
ICH Q2 (R1)、ICH Q7、ICH Q12 指导原则
#药品技术转移#分析方法转移#GMP合规#药企QA#药企质量控制#药企QC#生物制品#生物药方法转移
由于篇幅太长,本文配套的 6 份附录模板(方案 / 报告 / 豁免表 / OOS 处理流程 / 微生物模板 / 生物制品模板)已整理为 Word 版,可参考使用、按需取用。
领取方式:在公众号「李志红 LBC」,在主页 “发消息” 对话框回复关键词 方法转移模板,即可获取网盘链接与提取码。
