 夜雨聆风
夜雨聆风
Science: 合成超细单壁 MoS₂ 纳米管
随着集成电路器件尺寸不断逼近物理极限,如何获得原子级厚度且具有优异静电控制能力的沟道材料已成为下一代电子器件发展的核心问题。过渡金属硫族化合物(TMDs)纳米管兼具一维结构和半导体特性,其天然的圆柱形几何结构非常适合构建栅极全包围晶体管。此外,与碳纳米管不同,TMD纳米管不会因手性变化而出现金属性与半导体性的随机转换,因此被认为是极具潜力的新型沟道材料。理论研究早已预测,当MoS₂纳米管直径降低至数纳米甚至亚纳米尺度时,将出现丰富的新奇物理现象,包括手性依赖电子结构、可调带隙以及巨大的内建极化效应。然而受限于三原子层厚度MoS₂薄层极高的弯曲应变能,传统气相生长方法通常只能获得直径大于10 nm的多壁纳米管,难以实现小尺寸、单壁以及手性可控的结构。因此,如何突破尺寸和结构控制瓶颈,成为该领域长期面临的重要挑战。
据此, 东京大学Yusuke Nakanishi、国家先进工业科学技术研究院Ryosuke Senga、大阪大学Kazu Suenaga提出了一种基于氮化硼纳米管内部限域反应的新型合成策略。首先通过高真空蒸气反应将Mo₄S₄团簇填充进入开口的BN纳米管内部,随后在适当温度下退火,使这些团簇在受限空间中逐渐聚合并重构形成MoS₂纳米管。与传统生长方式不同,BN纳米管提供的一维纳米腔体不仅限制了前驱体运动自由度,同时能够通过范德华相互作用稳定高度弯曲的MoS₂结构,从而实现此前难以获得的超细单壁纳米管生长。
2026年6月4日, 相关工作以"Confined growth of armchair MoS₂ nanotubes at the 1-nm limit"为题发表在 Science 上。
图文介绍
通过高角环形暗场扫描透射电子显微镜(HAADF-STEM)和电子能量损失谱(EELS)分析,研究人员直接观察到了MoS₂纳米管嵌套于BN纳米管内部形成的同轴异质结构。元素映射结果清晰显示Mo和S元素仅分布于内层管体区域,证实了限域生长的成功实现。傅里叶变换分析进一步表明,所得结构保持六方晶格特征,与MoS₂晶体结构高度一致。这种由半导体MoS₂与绝缘BN构成的同轴纳米管异质结天然形成了栅极全包围器件所需的几何构型。
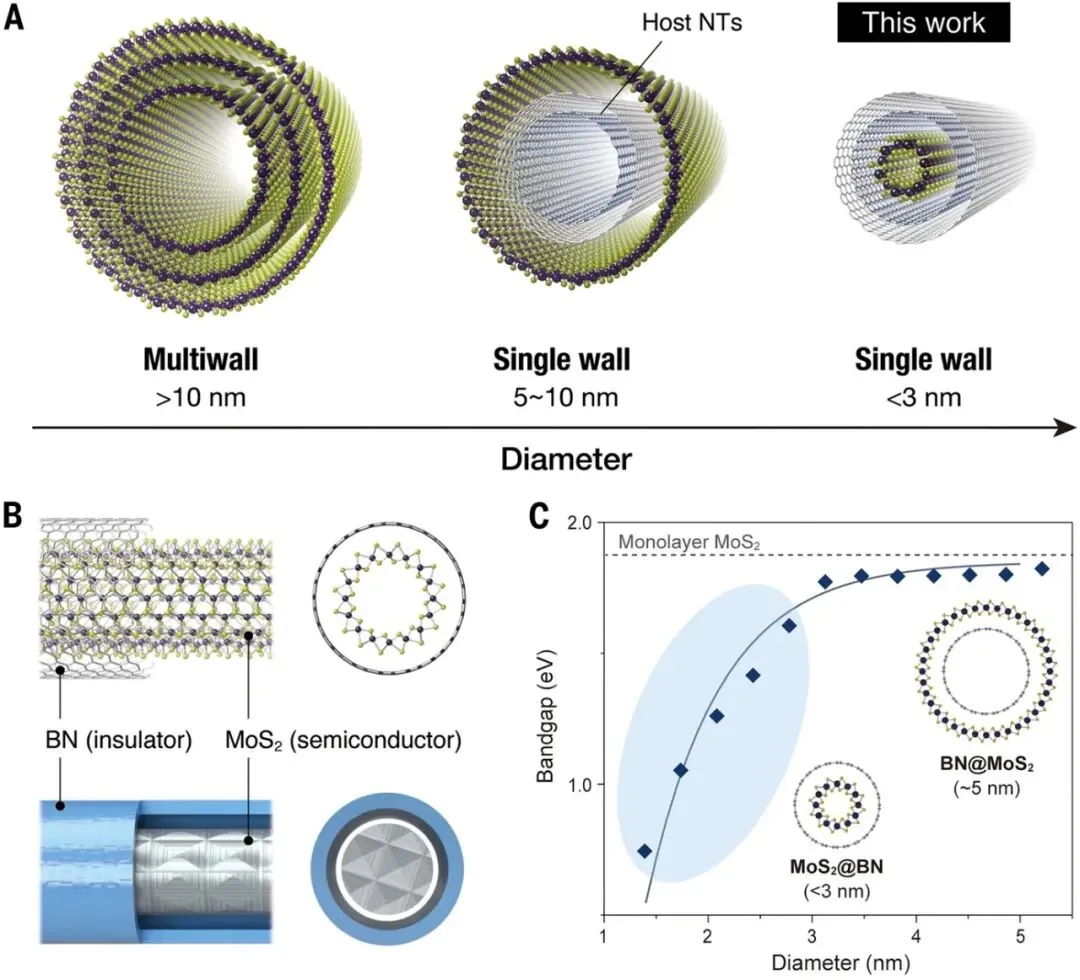 图1. 超细MoS₂纳米管的结构与电子特性。A)不同合成策略获得的MoS₂纳米管示意图:传统多壁纳米管、外壁生长单壁纳米管以及本工作实现的限域超细单壁纳米管。B)BN纳米管内部限域生长单壁MoS₂纳米管形成的同轴异质结构示意图。C)扶手椅型MoS₂纳米管带隙随直径变化关系。
图1. 超细MoS₂纳米管的结构与电子特性。A)不同合成策略获得的MoS₂纳米管示意图:传统多壁纳米管、外壁生长单壁纳米管以及本工作实现的限域超细单壁纳米管。B)BN纳米管内部限域生长单壁MoS₂纳米管形成的同轴异质结构示意图。C)扶手椅型MoS₂纳米管带隙随直径变化关系。
统计分析显示,所得MoS₂纳米管几乎全部为单壁结构,多壁纳米管出现概率极低。纳米管长度通常达到50–100 nm,部分样品可延伸至200 nm。更重要的是,其直径主要集中在2 nm以下,平均直径约为1.9 nm,最小直径接近1 nm。这一尺寸已经接近理论预测的MoS₂纳米管稳定存在极限。研究人员发现,MoS₂纳米管直径与BN纳米管最内层管壁直径之间呈现严格对应关系,两者之间保持约0.5 nm的层间距,与平面hBN/MoS₂异质结构中的范德华间距基本一致,表明BN纳米管主要通过弱相互作用稳定内层超细MoS₂结构,而不会显著改变其本征电子性质。
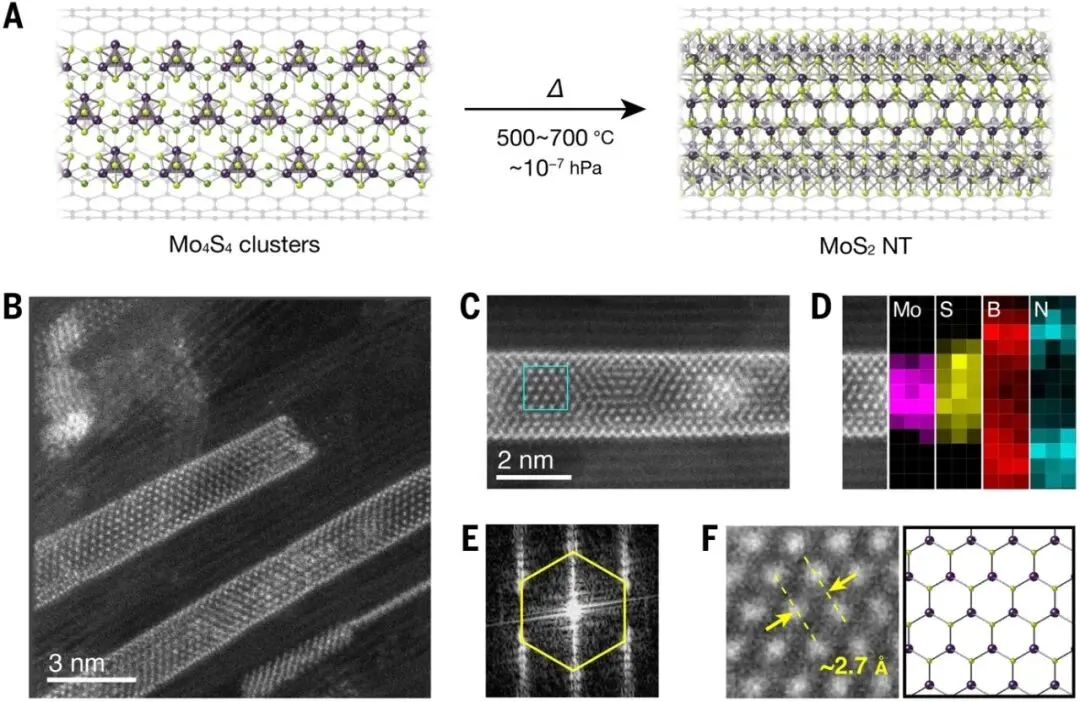
图2. BN纳米管内部单壁MoS₂纳米管的限域生长。A)Mo₄S₄团簇经退火转化为MoS₂纳米管的示意图。B)MoS₂纳米管嵌套于BN纳米管内部的HAADF-STEM图像。C)单根MoS₂@BN纳米管高分辨图像。D)对应的Mo和S元素EELS分布图。E)对应区域的傅里叶变换图样。F)局部放大图及其结构模型。
最引人注目的发现之一是超细MoS₂纳米管表现出异常强烈的扶手椅型(Armchair)手性选择性。对172根单壁MoS₂纳米管进行了系统统计,发现当直径低于约2 nm时,约84%的样品均为扶手椅型或接近扶手椅型结构,而传统外表面生长方法获得的MoS₂纳米管则呈现随机手性分布。进一步研究表明,这种手性选择性并非来源于BN纳米管模板的外延作用,因为宿主BN纳米管的手性与客体MoS₂纳米管之间不存在明显相关性。
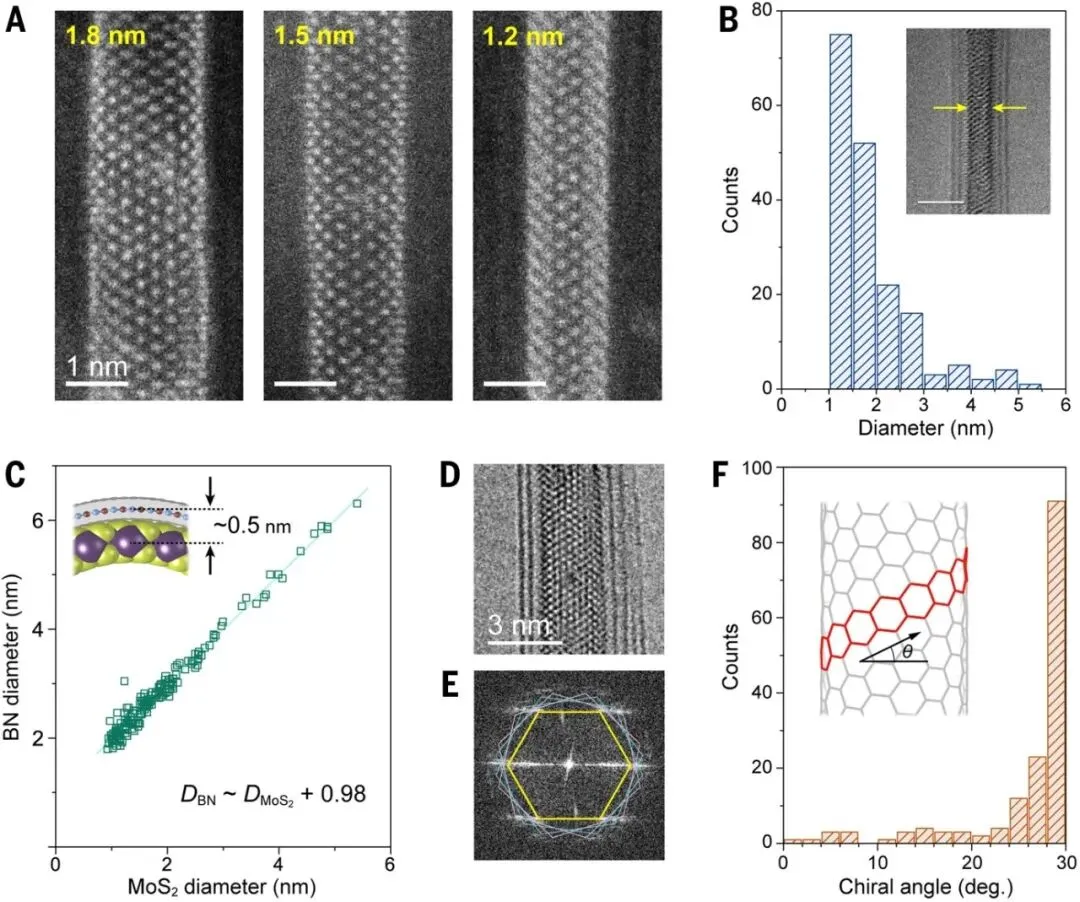
图3. 限域生长MoS₂纳米管的结构统计分析。A)不同直径超细单壁MoS₂纳米管的原子分辨图像。B)142根MoS₂纳米管的直径分布统计。C)MoS₂纳米管直径与BN纳米管内径之间的对应关系。D)单根MoS₂@BN纳米管TEM图像。E)对应傅里叶变换图样及手性分析。F)172根MoS₂纳米管的手性角分布统计。
为揭示手性选择性的形成机制,研究团队开展了第一性原理计算。结果显示,在约1.2 nm直径下,扶手椅型与锯齿型MoS₂纳米管具有几乎相同的总能量,因此实验观察到的扶手椅优势并非源于热力学稳定性差异。推测,在狭窄BN纳米管内部,前驱体团簇的运动受到严格限制,生长过程主要沿纳米管轴向进行,这种特殊的动力学环境更有利于形成扶手椅型结构。该结果说明,一维限域环境不仅能够控制尺寸,还可能直接决定最终形成的手性结构。
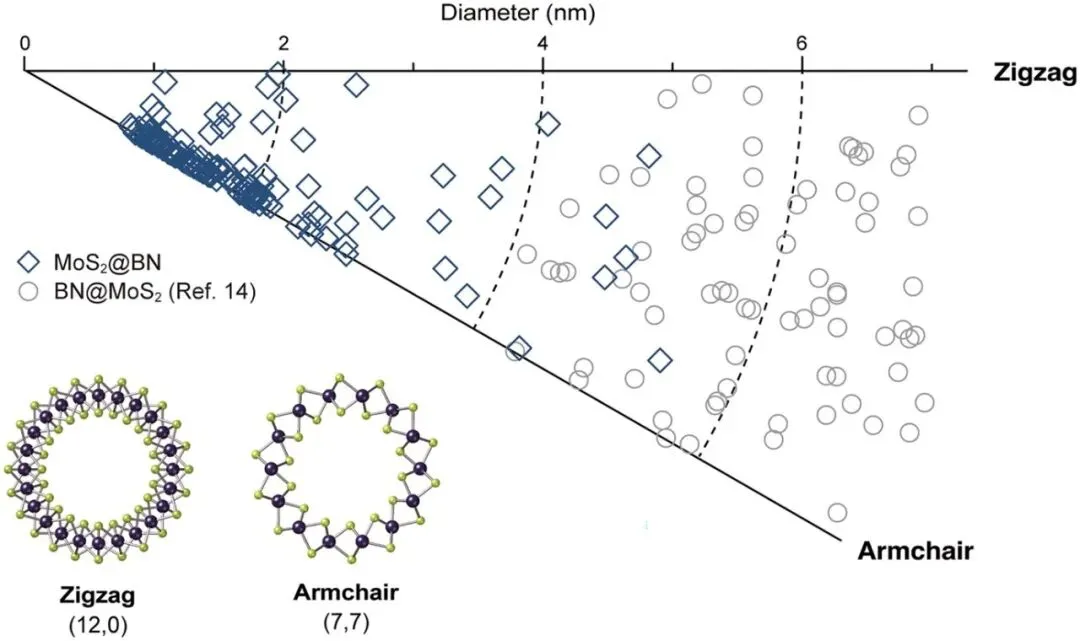
图4. 限域MoS₂纳米管的手性分布。不同直径MoS₂纳米管的手性分布图,以及典型锯齿型和扶手椅型结构模型。
在电子结构研究方面,研究团队利用单色化低损失EELS测量了不同直径MoS₂纳米管的光学响应。实验发现,随着直径从约3 nm进一步减小至1 nm量级,光学带隙从接近单层MoS₂的1.8 eV逐渐降低至约1.3–1.5 eV。该结果首次在实验上证实了理论预测多年的“曲率诱导带隙收缩效应”。与碳纳米管中常见的带隙增大趋势不同,MoS₂纳米管由于强曲率引起Mo-d轨道与S-p轨道杂化状态发生重构,从而导致价带顶上移、导带底下移,最终表现为带隙持续减小。这种结构与电子性质之间的直接关联,为未来通过几何尺寸精确调控半导体性能提供了新的策略。
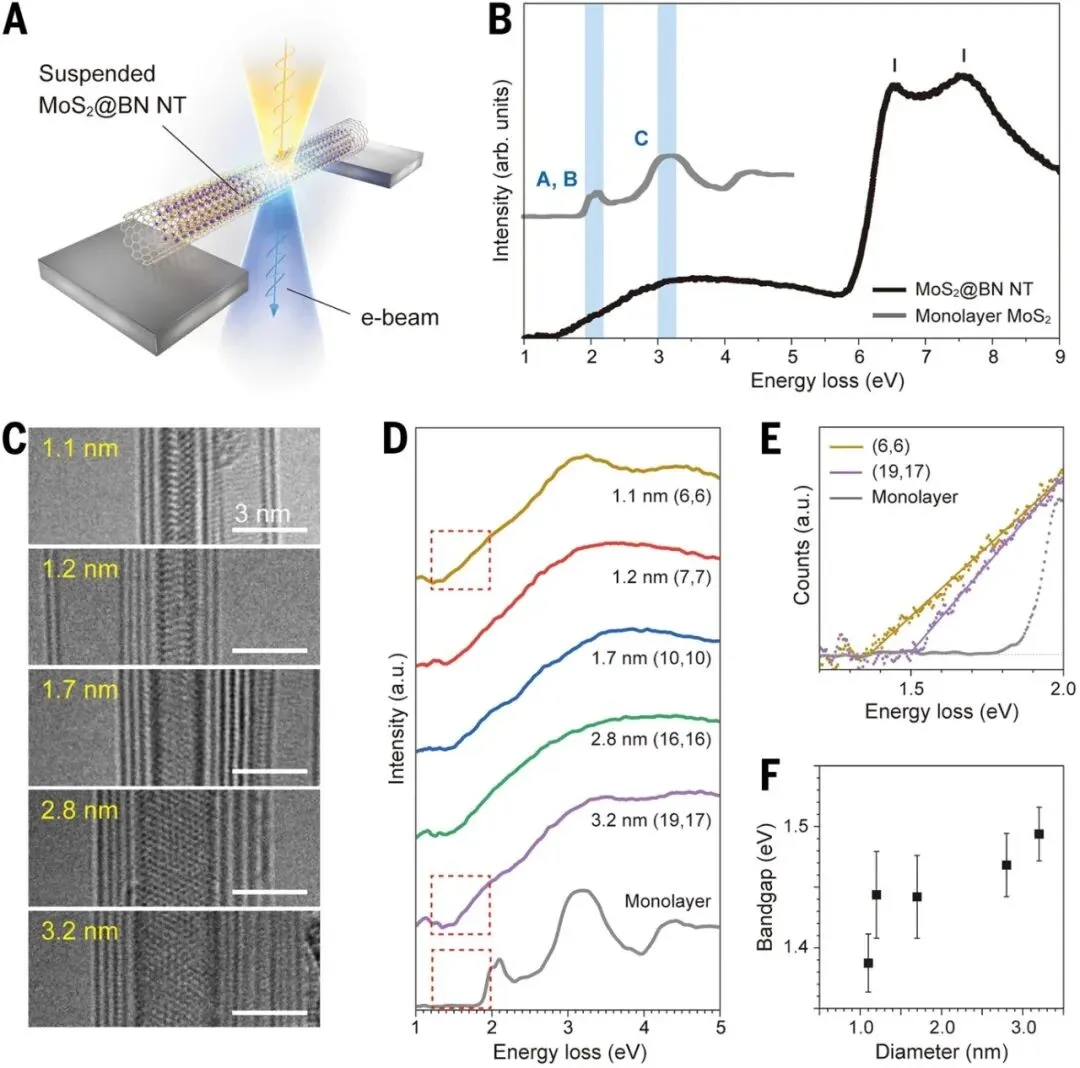
图5. 超细MoS₂纳米管的尺寸依赖光学性质。A)单色化STEM-EELS测试示意图。B)1.6 nm MoS₂纳米管与单层MoS₂的低损失EELS谱对比。C)不同直径MoS₂纳米管TEM图像。D)对应低损失EELS谱。E)不同结构MoS₂纳米管吸收边比较。F)MoS₂纳米管光学带隙随直径变化关系。
综上, 首次实现了直径接近1 nm极限的单壁MoS₂纳米管可控制备,并揭示了一维限域环境下的尺寸调控、手性选择以及电子结构演化规律。氮化硼纳米管不仅作为限域模板稳定了高曲率MoS₂结构,同时天然形成绝缘包覆层,为构筑原子级栅极全包围晶体管提供了理想平台。研究成果表明,限域生长策略有望推广至其他过渡金属硫族化合物以及层状材料体系,为构建原子级精度的一维范德华异质结构开辟新路径。随着相关合成技术和器件加工工艺进一步成熟,这类超细半导体纳米管有望在未来超高密度集成电路、量子电子学、光电子器件以及新型低功耗计算架构中发挥重要作用。
原文链接
https://www.science.org/doi/10.1126/science.aee3446
Science:光催化氧化裂解烯烃
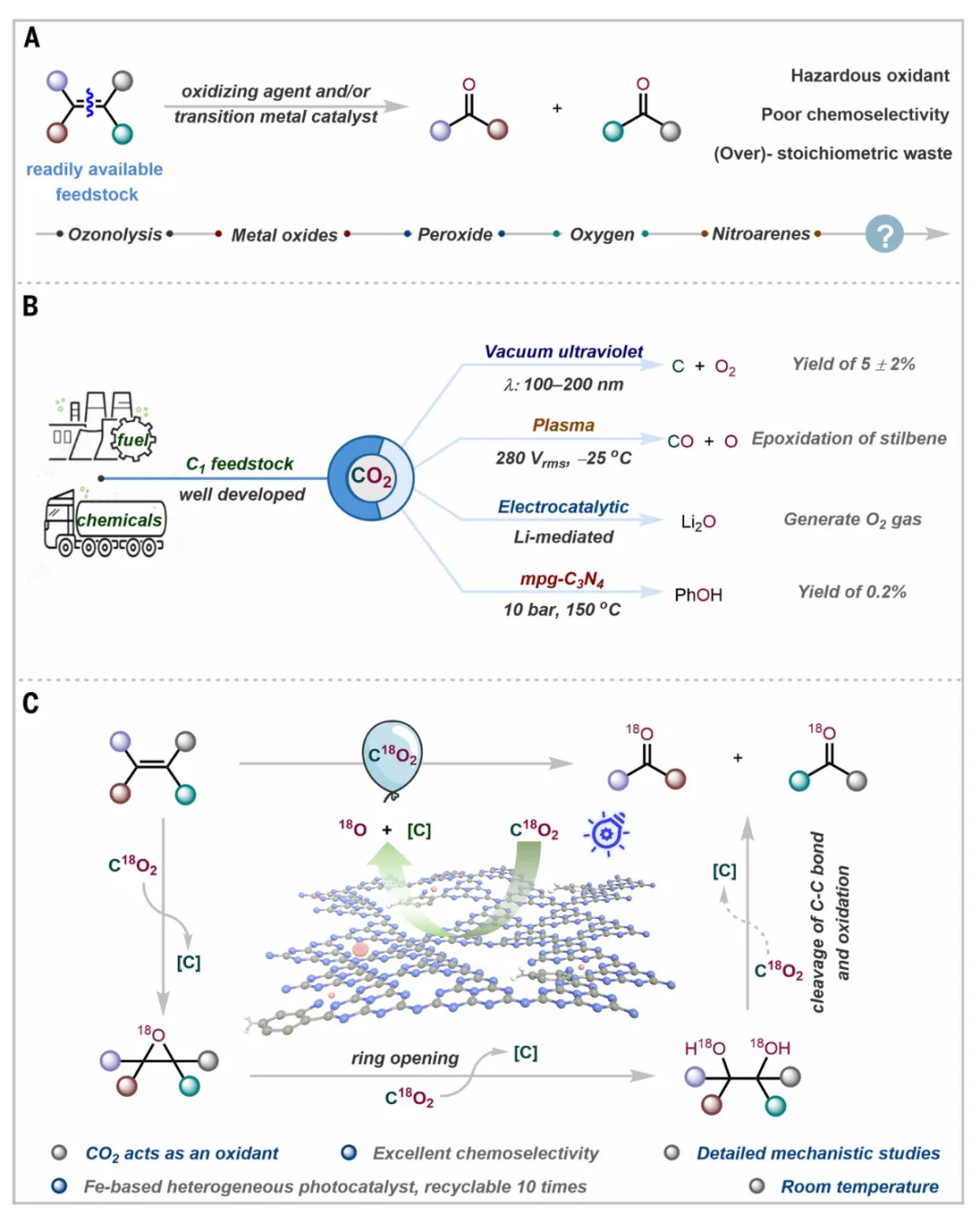
氧化裂解碳碳双键是有机合成中最重要且应用最广泛的转化之一,其产物在化学工业中占比近30%。传统的烯烃氧化裂解方法主要依赖臭氧分解、Lemieux-Johnson氧化或锇基试剂等,这些方法通常需要使用化学计量甚至过量的有毒、易爆或高污染试剂(如臭氧、高锰酸盐、四氧化锇),不仅产生大量废弃物,还带来严重的安全隐患。尽管分子氧作为绿色氧化剂具有一定优势,但富氧环境加剧了易燃风险,限制了其在工业规模上的应用。近年来,硝基芳烃光氧化裂解方法虽有所发展,但仍需使用化学计量的硝基化合物,产生等摩尔废弃物。因此,开发一种使用绿色、安全氧化剂且条件温和的氧化裂解策略,是现代合成化学中亟待解决的问题。二氧化碳作为一种无毒、安全、廉价的C₁原料,其氧含量丰富,若能在温和条件下有效利用其氧原子,将有望成为有机合成中最绿色的氧化剂之一。然而,CO₂分子具有极高的热力学稳定性(ΔG°≈-94.6 kcal mol⁻¹)和极强的C=O键(键解离能≈179 kcal mol⁻¹),传统上很难将其作为氧供体使用。
据此,德国拜罗伊特大学Shoubhik Das、莱布尼茨催化研究所Matthias Beller报道了一种光催化策略,利用温和的二氧化碳(CO₂)作为氧供体,在常温常压下将烯烃转化为酮或羧酸。一种坚固的铁基异质光催化剂促进了氧转移,形成环氧化物中间体,随后进行开环和碳-碳键断裂,以高选择性获得氧化产物。全面的机理研究结合了时间分辨光谱、同位素标记和原位光谱分析以及先进的量子力学模拟。这些结果揭示了光催化条件下从CO₂进行氧转移的基本原理,为光驱动的绿色氧化转化提供了一个可持续的平台。
2026年6月4日, 相关工作以"Photocatalyzed oxidative cleavage of alkenes using CO₂ as an oxygen donor"为题发表在 Science上。
图文介绍
清晰地将传统烯烃氧化裂解方法与本文发展的CO₂氧供体策略进行了系统对比。传统方法中,臭氧分解虽然应用广泛,但臭氧具有剧毒和爆炸性,且需要化学计量使用,带来严重的安全问题。Lemieux-Johnson氧化和锇基方法则依赖重金属,产生大量有毒废弃物。即使采用分子氧作为氧化剂,富氧环境也显著增加了火灾和爆炸风险,限制了其大规模工业应用。近年来发展的硝基芳烃光氧化方法虽然条件温和,但仍需使用化学计量的硝基化合物,废弃物问题依然存在。与之形成鲜明对比的是,本文提出的策略直接使用CO₂作为氧源,CO₂不仅无毒、不可燃,而且是极度丰富的温室气体,将其转化为有价值的化学品具有环境和经济的双重意义。还展示了本工作的核心创新,即通过铁基异相光催化剂Fe@f-gC₃N₄,在可见光照射下激活线性CO₂分子,使其弯曲变形并发生C=O键断裂,从而产生活性氧物种,这些物种随后将氧原子转移到烯烃的C=C双键上,最终生成酮或羧酸。这一设计巧妙地将CO₂从传统的C₁资源扩展为一种新型的氧供体,为光催化氧化开辟了全新的方向。
该光催化氧化裂解方法对多种烯烃底物的适用性,共包含超过45个示例,产率从41%到90%不等。对于1,2-二取代芳基烯烃,无论芳环上带有给电子基团(如甲氧基、甲基)还是吸电子基团(如三氟甲基、氯、硝基),均能以良好至优异的产率得到相应的苯甲酸产物,体现了对电子效应的宽适应性。值得注意的是,该方法对醛基、羟基、炔基以及Boc保护的胺基等通常对强氧化条件敏感的官能团表现出高兼容性,例如含有醛基的底物2ac在0°C下仍能顺利反应且醛基本身未被氧化,这凸显了该催化体系的温和性与选择性。对于环状烯烃(如环己烯、环戊烯),反应同样顺利进行,生成相应的二酸或酮酸产物。此外,该策略成功拓展至非活化的脂肪族烯烃,包括1-辛烯和1-癸烯,分别以中等产率得到庚酸和壬酸,证明了该方法对非共轭烯烃同样有效。更重要的是,图2还展示了该方法在复杂天然产物和药物分子后期修饰中的应用潜力,例如由非诺贝特、氟哌啶醇、多奈哌齐、雌酮等衍生的烯烃底物均能以55%-79%的产率实现氧化裂解,得到相应的多官能化酮或羧酸产物。这些结果强有力地证明了该光催化体系在合成化学和药物化学中的实用价值。
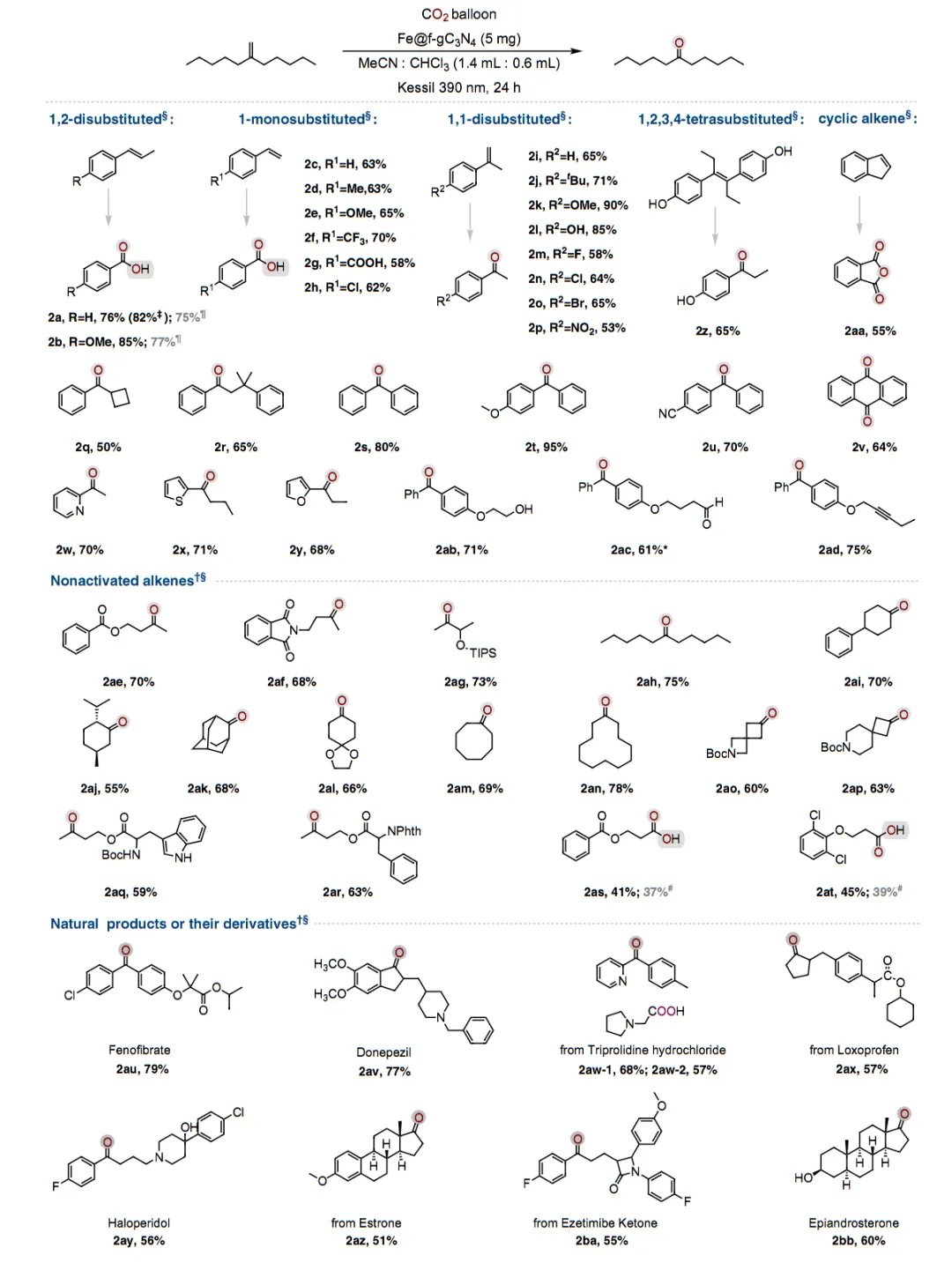
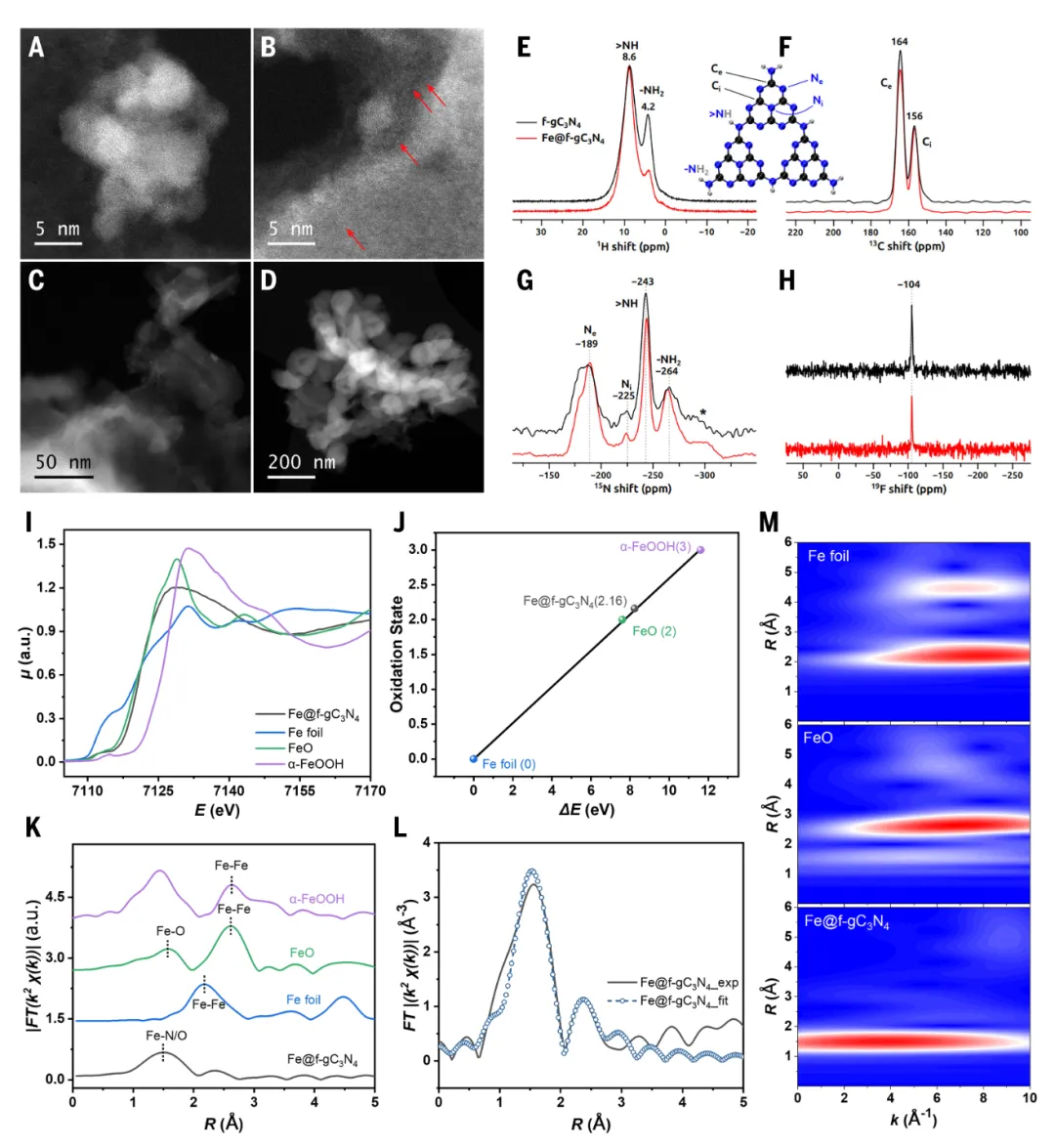
通过多种先进表征手段对催化剂Fe@f-gC₃N₄的结构进行了深入解析。高角度环形暗场扫描透射电子显微镜图像清晰地显示出铁物种在改性氮化碳载体上的分布状态,其中既有单原子分散的铁位点(红色箭头标示),也有纳米尺度的氧化铁颗粒,这种原子级分散与纳米颗粒共存的混合状态可能是催化剂高活性的关键。X射线光电子能谱分析表明,催化剂表面存在FeOₓ和金属态Fe两种物种,且金属态与氧化态的比例约为0.37,铁主要以+2价存在。固态核磁共振波谱进一步揭示了载体的化学结构,其中¹⁹F魔角旋转核磁共振在-104 ppm处出现特征信号,结合密度泛函理论计算,表明引入的含氟前体在催化剂制备过程中发生了部分脱氟,最终以=CF₂或-CHF₂的形式存在于材料中,而非原始的-CF₃基团。X射线吸收精细结构谱提供了铁局域配位环境的关键信息。Fe K边X射线吸收近边结构分析表明,反应前后催化剂中铁的氧化态均为约+2.16,证明催化剂在循环过程中保持了结构稳定性。扩展X射线吸收精细结构拟合结果显示,Fe的配位环境以Fe–O/N键为主,键长为2.03 Å,配位数远低于FeO标准样品,这与单原子铁和氧化铁纳米颗粒共存的结论一致。反应后样品中Fe-Fe散射路径的增强表明有部分铁物种发生了聚集,但整体催化性能并未显著下降,体现了材料的稳健性。
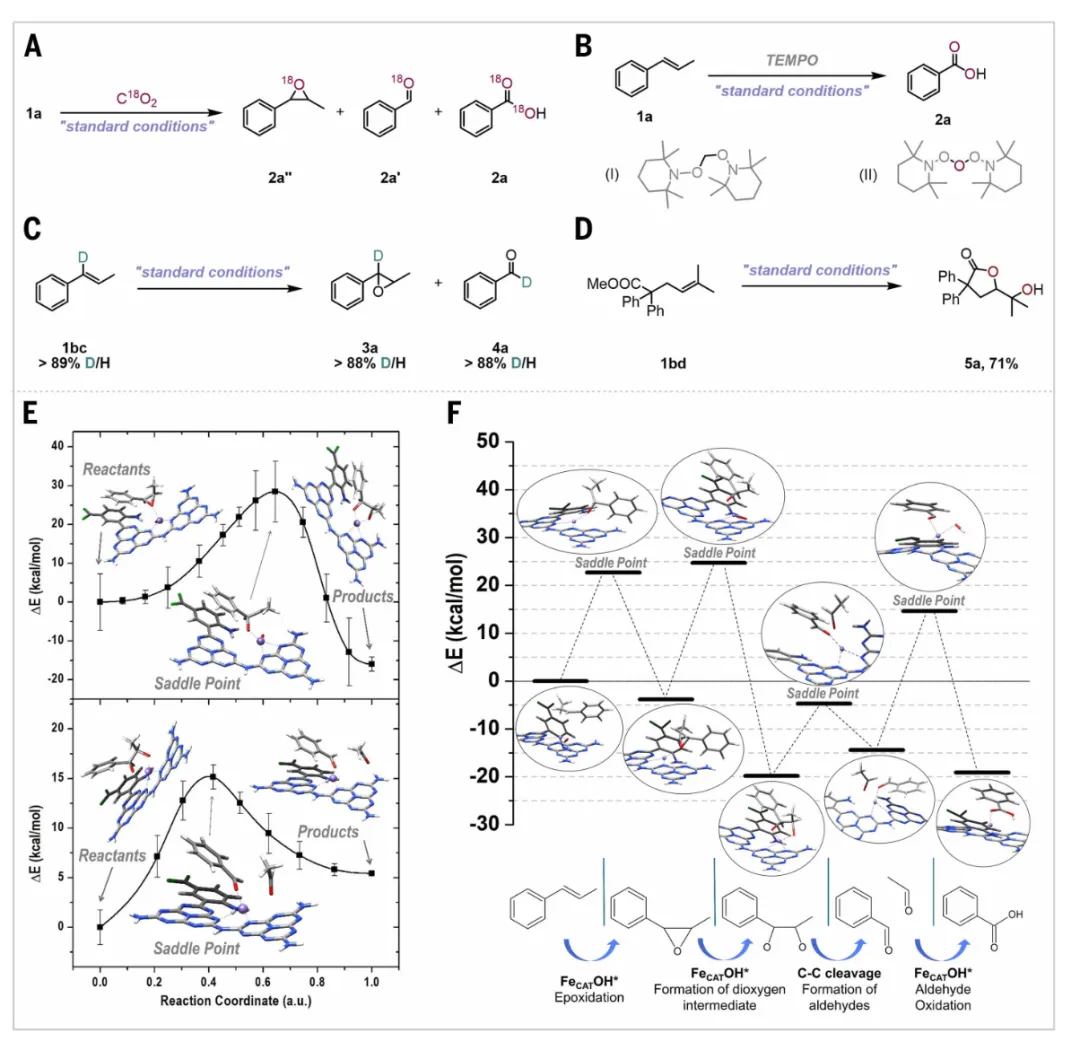
通过一系列精巧的实验和理论计算,阐明了CO₂作为氧供体实现烯烃氧化裂解的反应路径。首先,C¹⁸O₂同位素标记实验提供了最直接的证据,当反应在C¹⁸O₂气氛下进行时,所生成的环氧化物、苯甲醛和苯甲酸中均检测到了¹⁸O的掺入,且随时间演化规律一致,证实在整个反应过程中所有含氧化合物的氧原子均唯一来源于CO₂,而非来自溶剂或空气。中间体捕获实验成功检测到了由CO₂衍生的活性氧物种,高分辨质谱中观测到Fe-O-O-H等关键中间体的特征峰,同时原位红外光谱检测到了表面吸附的CO₂弯曲振动模式,证实了CO₂在铁位点上发生了线性到弯曲的构型转变,从而降低了C=O键活化的能垒。自由基淬灭实验表明,加入TEMPO后反应完全被抑制,表明该反应涉及自由基途径。环丙烷基自由基钟底物(如1,1-二苯基乙烯)在标准条件下生成了环丙烷开环产物,进一步支持了自由基中间体的参与。密度泛函理论计算详细模拟了从CO₂活化到最终产物生成的全过程自由能变化。计算表明,CO₂在Fe位点上化学吸附并弯曲后,在质子辅助下发生C-O键断裂,生成Fe=O活性物种,该物种与烯烃配位后形成环氧化物中间体,环氧化物开环生成二醇,随后C-C键断裂生成醛,最后醛被进一步氧化为羧酸。整个过程的速控步为环氧化物的开环步骤,能垒约为20 kcal mol⁻¹,与温和的实验条件相吻合。这些系统的机理研究不仅解释了实验现象,也为设计更高效的CO₂氧供体光催化剂提供了理论基础。
总之, 通过创新的光催化策略,成功解决了利用热力学极稳定的二氧化碳作为温和氧化剂的科学难题。利用精心设计的铁基异质催化剂,不仅实现了在常温常压下对烯烃C=C键的选择性氧化断裂,更通过详尽的机理研究阐明了CO₂活化与氧转移的微观过程。这一成果不仅为有机合成提供了一种比臭氧分解更安全、环保的替代方案,也为CO₂的捕集与资源化利用开辟了全新的高附加值应用路径
投稿、转载、合作请联系微信:MatResFron001
《电介质Dielectrics》微信公众号诚邀各位读者投稿,投稿方向包含但不局限于:近期科研成果介绍、经典文章深入解读、测试原理介绍、科研软件分析、软件操作教程类、科研入门类教程、相关书籍推介、科研技巧分享、学术会议讲座推荐、以及招生招聘等。

为作者打call,点击右下角“赞”和“在看”↓↓↓
