 夜雨聆风
夜雨聆风





医疗器械设计开发文档
↑请关注公众号【医械人学习圈】,欢迎转发
在2025年国家药监局公开发布的《医疗器械生产质量管理规范》(2025年第107号)中,第七章设计开发章节中的第五十七条关于设计开发文档的,条款具体为:企业应当建立产品设计开发文档,包括设计开发过程中建立或者引用的记录,确保历次设计开发输出过程以及相关活动可追溯。
由此可见,对于医疗器械的设计开发文档,法规的要求也在进一步提高。
一、 首先来看看各国对于医疗器械设计开发文档的要求
二、 什么是设计开发文档
a)国家GMP定义设计开发文档为医疗器械的设计开发过程中建立或者引用的记录。
b)DHF (Design History File),是记录医疗器械是如何设计的。即FDA将设计开发文档定义为“描述成品医疗器械设计开发历史的记录汇编”。
所以,设计开发文档就是医疗器械在设计过程中所产生的所有记录的汇编。
三、 为什么需要设计开发文档
-
满足法规合规要求各国监管机构(如中国NMPA、美国FDA、欧盟CE等)均要求医疗器械企业建立完整的设计开发文档,以证明产品设计符合安全、有效性和质量可控性要求。例如,中国《医疗器械生产质量管理规范》明确要求设计开发文档需涵盖设计输入、输出、验证、确认等全过程记录,确保产品全生命周期合规。
-
确保产品质量与安全性设计开发文档记录了产品从需求定义、设计输入到验证确认的全过程,有助于系统性地识别和控制风险。通过文档化的风险管理措施、性能测试数据等,可确保产品在设计阶段就满足安全性和有效性要求,减少因设计缺陷导致的安全事故。
-
支持注册申报与审评注册申报资料(如产品技术要求、临床评价报告、风险管理文件等)均来源于设计开发文档。完整的设计开发文档为注册申报提供了充分的技术依据,有助于提高审评效率,降低注册失败风险。
-
实现过程可追溯性设计开发文档记录了产品的设计变更、验证活动、评审记录等信息,便于在产品生命周期内追溯设计决策的依据和过程。当出现质量问题或需要进行产品改进时,可通过文档快速定位问题根源,采取有效措施。
-
促进团队协作与知识传承医疗器械研发涉及多学科团队(如机械、电子、软件、临床等),设计开发文档作为共同的语言,有助于团队成员理解产品设计目标、技术要求和工作进展,促进跨部门协作。同时,文档也是企业知识资产的重要组成部分,为新项目开发提供参考,避免重复犯错。
-
应对市场与监管变化医疗器械行业法规和标准不断更新,设计开发文档可帮助企业快速响应变化。通过文档记录的设计输入、风险控制措施等,可评估法规变化对产品的影响,及时调整设计和开发策略,确保产品持续合规。
四、 设计开发文档包括哪些内容
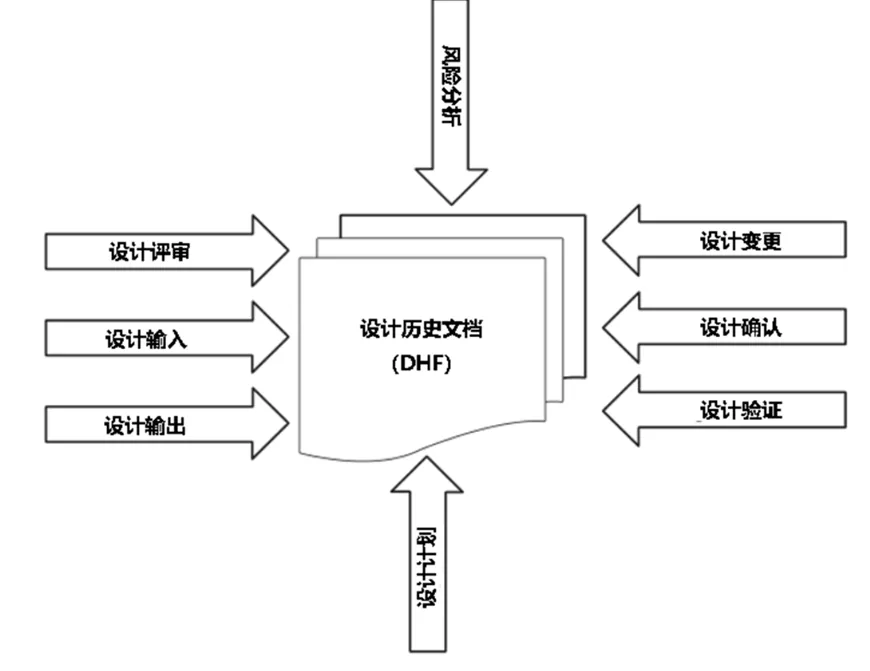
根据图片上的要求,设计开发文档包含了医疗器械产品设计开发的所有过程所产生的记录,也包括设计开发计划和过程中所有的风险管理的记录。
往期精选
宣贯官方免费回放《医疗器械生产质量管理规范》,错过直播的看过来!
宣贯《医疗器械生产质量管理规范》国家局11月17日李处长宣讲视频PPT


医械人学习圈
关注更多精彩内容
