 夜雨聆风
夜雨聆风





HiMT:植物线粒体/叶绿体自动化组装神器来啦!
植物线粒体与叶绿体基因组在基因功能解析、进化研究和定向育种中具有重要价值,但其组装受频繁重组和大量重复序列影响而困难重重。
近日,中国热带农业科学院热带作物品种资源研究所、热带作物生物育种全国重点实验室团队在《Plant Communications》上发布跨平台细胞器基因组研究软件——HiMT,重点关注植物线粒体基因组,集成组装、评估、比较和可视化等模块,支持了细胞器基因组的大规模低成本高效率一键自动化组装。

植物线粒体基因组重复序列多、重组事件频繁,其高效组装存在明显挑战。为此,作者开发了 HiMT(High-fidelity data-based Mitochondrial genome assembly Toolkit)软件。其支持默认参数下的一键式叶绿体和线粒体基因组组装、可跨平台使用、 具有用户友好界面,并可生成用于细胞器基因组组装质量评估的交互式报告。HiMT在保持低硬件需求和少运行时间的同时,确保了组装的高完整性和高成功率。该软件免费向用户开放,
在线网址:https://github.com/tang-shuyuan/HiMT
英文手册:https://www.yuque.com/yuqueyonghuwrgkbo/tonqgq
中文手册:https://www.yuque.com/yuqueyonghuwrgkbo/aeg17o/ifmwlcuydrk3m35r
HiMT算法与逻辑设计
在植物全基因组测序中,细胞器基因组读段深度通常远高于核基因组。基于这一特征,HiMT首先利用长度长数据,通过保守的24个线粒体蛋白编码基因评估线粒体保守基因的测序深度,筛选含高频k-mer的细胞器基因组读段(图1)。采用优化的Kmer存储和统计模式,极大了节省计算资源(约为传统模式的1/64)。进一步通过图形组装工具(如Flye)高效完成细胞器基因组组装。
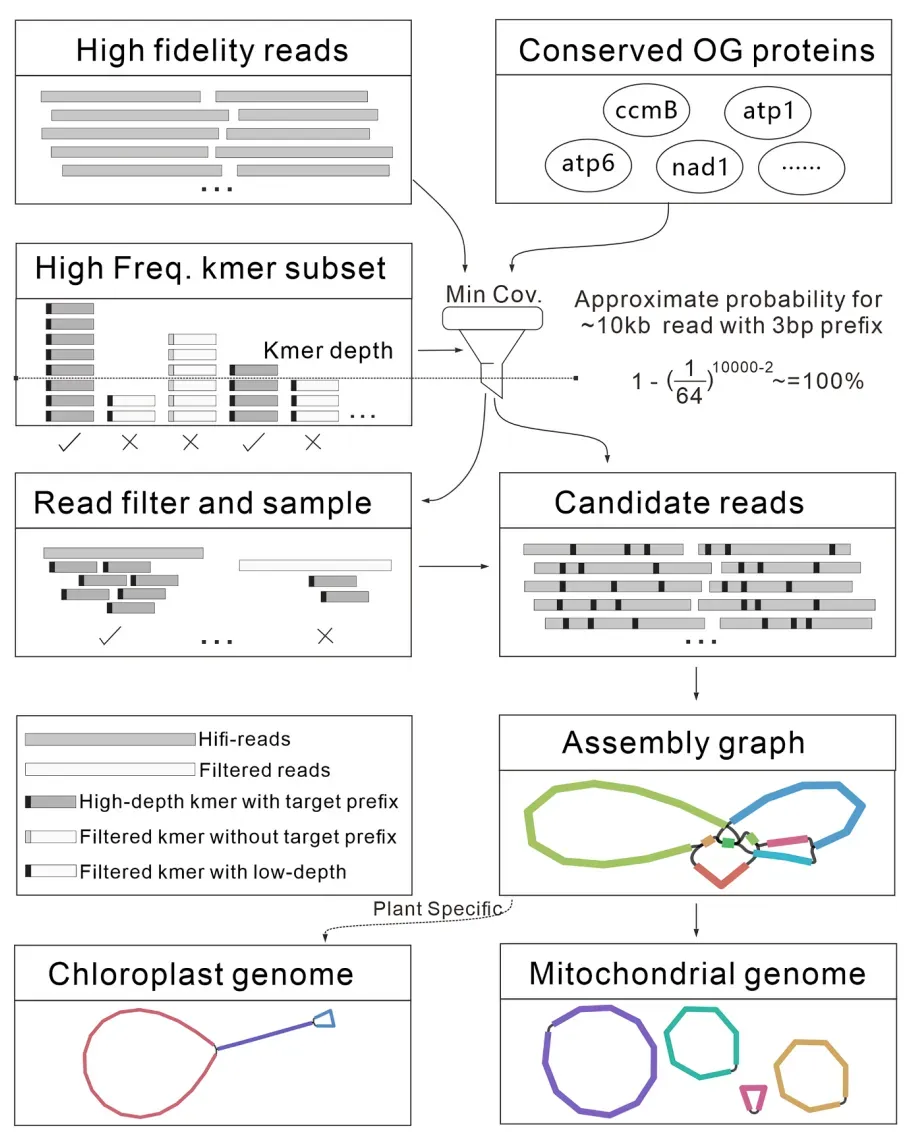
图 HiMT算法优化与流程设计总览
利用 HiFi 读段与保守细胞器蛋白序列自动确定含 3-bp 前缀的高频 k-mer 阈值,识别候选读段后分别组装叶绿体与线粒体基因组。
HiMT功能概览
HiMT 将核心功能拆分为“组装、读段过滤、质量评估、结果比较”四大模块(图 2A),并提供命令行一键模式(图 2B)和基于 TBtools-II 的图形界面(图 2C)。
通过对多个依赖软件包的代码进行了重构,HiMT具备完全的跨平台兼容性。该软件主程序包含四大核心功能如下:
● 用于细胞器基因组组装的”Assembly”模块;
● 独立读段过滤的”Read Filter”模块;
● 评估组装质量的”Quality Assess”模块;
● 比较不同线粒体组装结果的”Compare”模块。

图 HiMT功能和图形用户界面(GUI)概述
(A) HiMT 三大软件功能与四种操作模式。(B)命令行一键组装示例及输出文件(mt/cp 分别代表线粒体/叶绿体)。(C)图形用户界面演示。(D)交互式组装质量报告示例。
HiMT组装性能
以菠萝品种 ‘BL’ 和水稻为例,将 HiMT 与现有工具 PMAT 进行全面对比。菠萝线粒体基因组(911,652 bp)两工具长度完全一致,ANI 高达 99.99%;水稻 454 kb 线粒体基因组结果同样 100% 一致。进一步对 24 种植物 26 份样本的测试表明,HiMT与PMAT组装长度差异极小,ANI 介于 99.95–100%,证明HiMT在完整性上与PMAT等效。
此外,对于有经验的用户,可借助HiMT的”Read Filter”功能,整合HiMT的读段过滤算法和其他软件如PMAT/Canu等的组装算法,实现自定义的高效一键自动化组装流程。
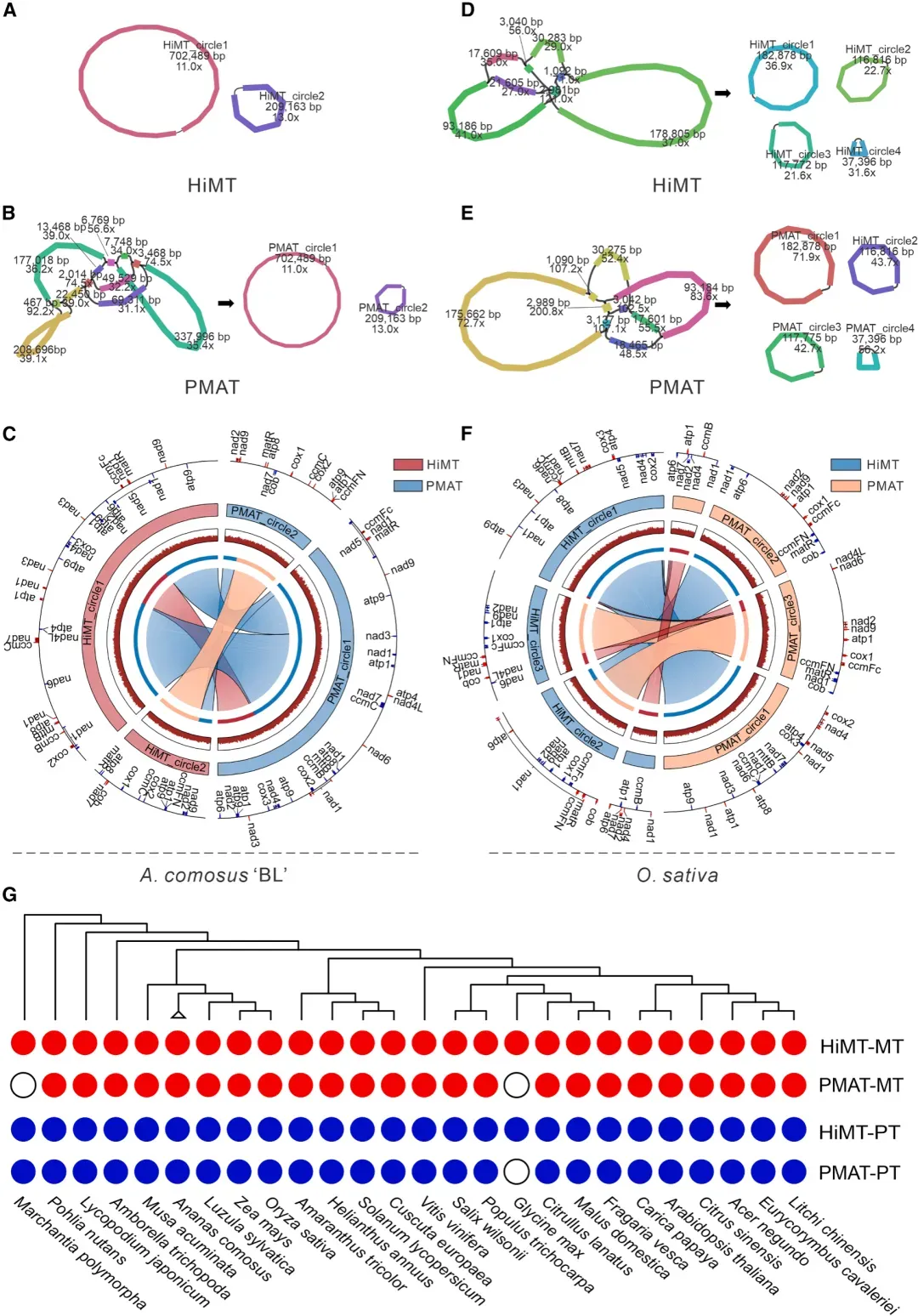
图 HiMT组装结果评估
(A-C)采用HiMT与PMAT对菠萝线粒体基因组进行的组装结果对比分析。(A)和(B)为通过Bandage生成的组装可视化图,(C)为采用Circos软件绘制的基因组对比环形图。
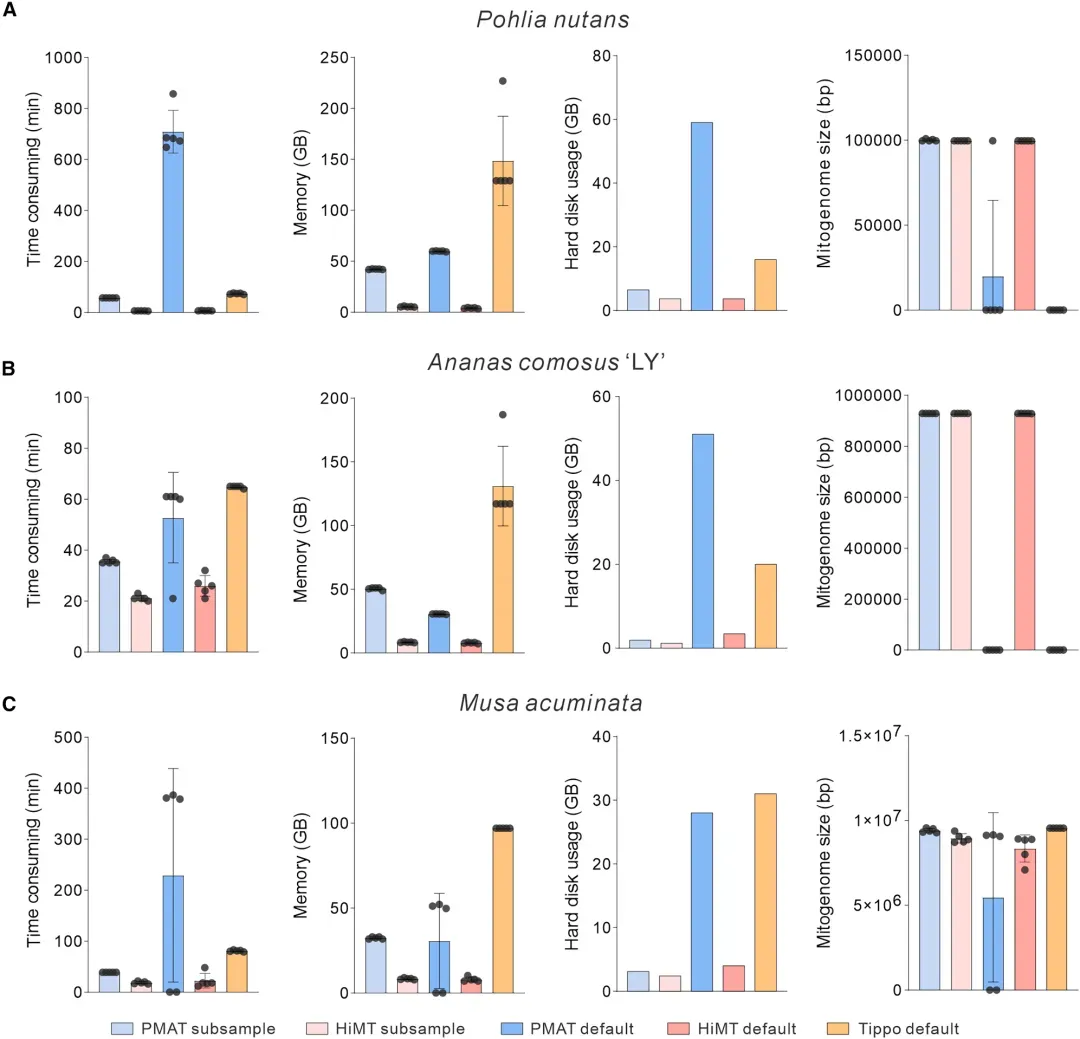
图 HiMT、PMA与TIPPo组装效率对比
选取黄丝瓜藓(~100 kb)、凤梨(~1 Mb)及小果野蕉(>10 Mb)三类代表性数据集,比较 HiMT、PMAT、TIPPo在默认与采样模式下的资源消耗,说明HiMT在笔记本级别硬件上的高效性与实用性
交互式组装质量评估报告
针对植物线粒体基因组评估工具的缺失现状,HiMT开发了”Assess”评估模块。该功能可直接解析GFA/FASTA格式的线粒体基因组组装结果,生成包含核心质量指标的交互式HTML报告。此外,若存在近缘物种或同物种已有组装结果,HiMT可进行快速比较分析,自动生成全局比对图谱,显示基因组序列异同。
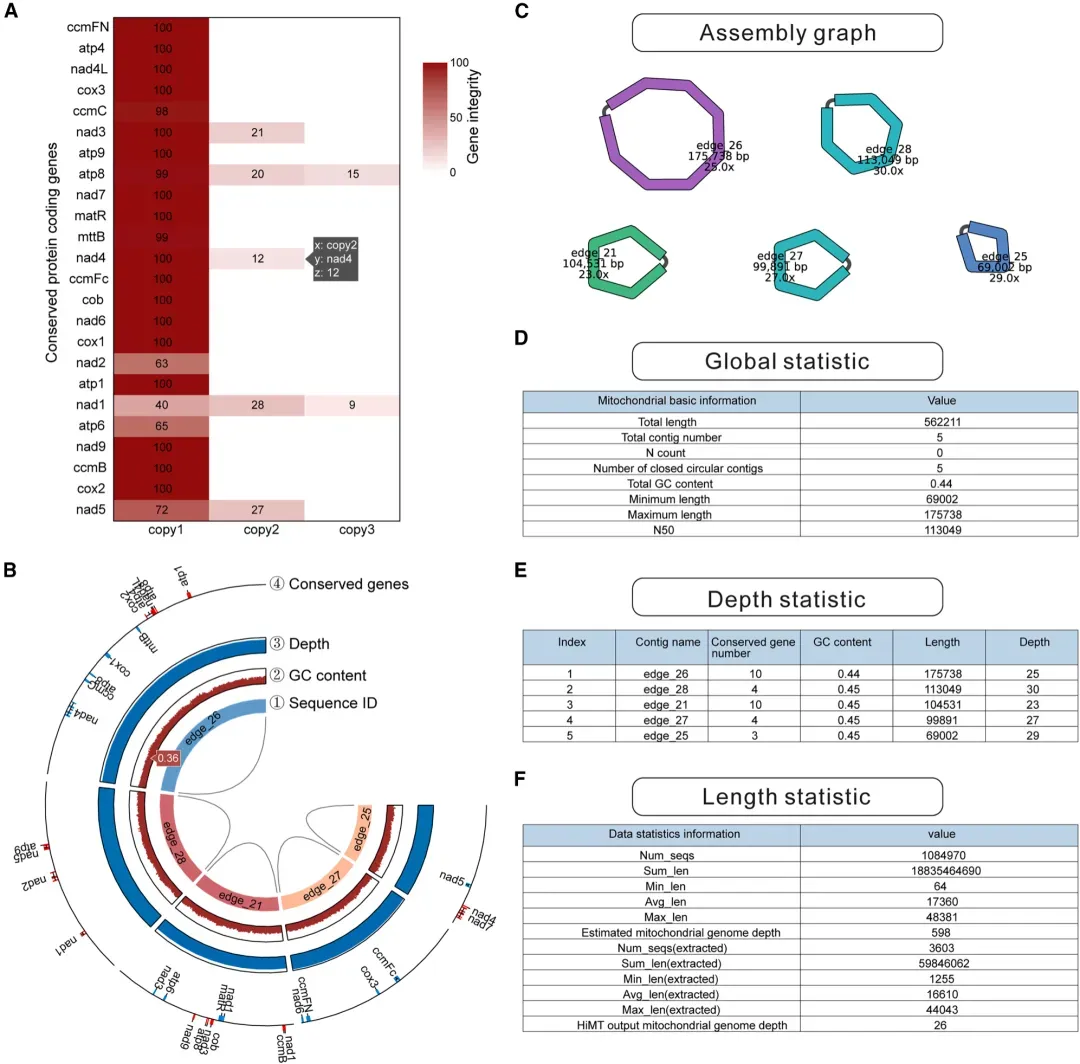
图 植物线粒体基因组组装完整性评估报告示例
(A) 保守基因序列相似度交互热图。 (B)交互式 Circos:由内到外依次为 contig ID、GC 含量、覆盖 深度、保守基因位置。(C–F) 分别为基因组长度、GC、N50、深度及输入/过滤数据统计表。
总结
研究通过采用基于测序深度估计和简化k-mer频率分析的简洁高效策略,开发了HiMT软件,显著提升了组装效率,大幅缩短计算时间,有效降了低资源消耗,且具备用户友好特性,对推动植物线粒体基因组研究具有重要价值。未来将优化超大线粒体(>10 Mb)组装,并增强对结构变异的解析能力。



凌波微课的讲师们,实战经验丰富,旨在通过在线微课程及线下交流,帮助科研学生及科研工作者们由简入繁,掌握科研思路及生信分析的实际操作。倾心打造的高通量测序数据分析平台——凌波微课·云平台(http://www.cloud.biomicroclass.com/CloudPlatform/home),无需命令行,零基础玩转数据分析。凌波微课,用心服务科研用户,打造专业教育品牌,助力科研提升。

关注我
发现更多精彩
关注凌波微课公众号,回复“入群”,即可加入凌波微课课下交流群,更多干货等你呦!