 夜雨聆风
夜雨聆风





AI多肽生成工具LigandForge很惊艳,覆盖150个受体靶点,预测与实验亲和力的相关性高达83%,比Bindcraft和Boltzgen快10000倍至100万倍
能以比Bindcraft和Boltzgen等最先进方法快10,000倍至100万倍的速度生成高质量肽段。
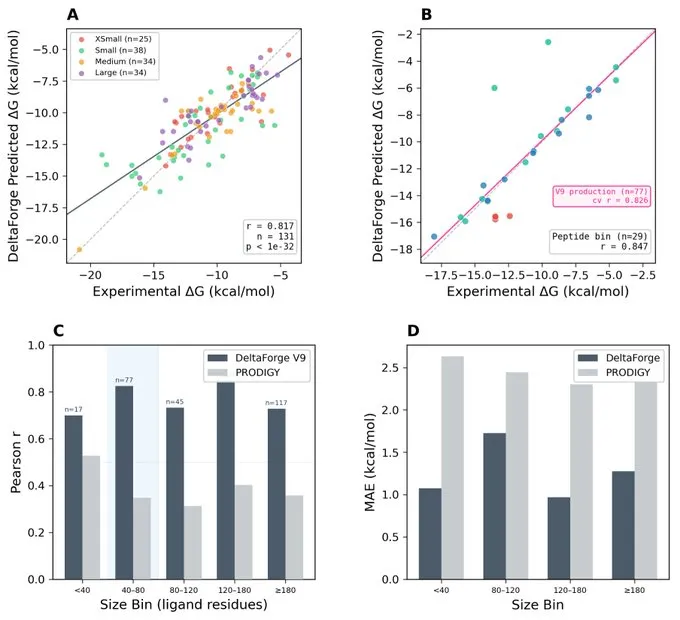
预测结合亲和力与实验结合数据的相关性高达83%。已对150个蛋白质靶标进行了基准测试。
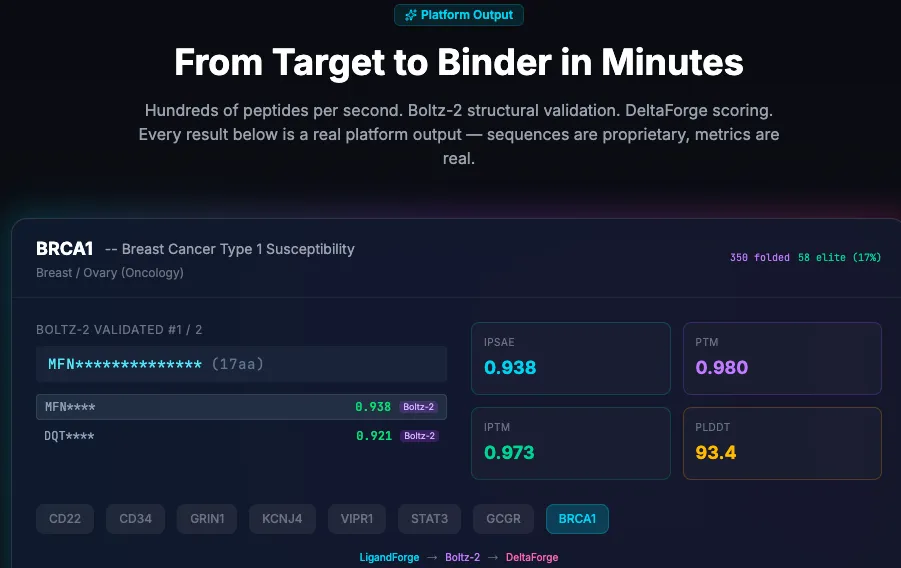
针对150个蛋白质和受体靶点生成了 490,691个肽段,并通过Boltz-2结构预测验证了其中16,475 个。
与 Bindcraft 和 Boltzgen 直接比较时,作者的分层监督离散扩散神经网络在界面质量和计算评分结合能方面超越了最先进的基准,同时生成肽的速度比每种方法快 10,000 倍 – 100 倍以上。
这意味着什么?在设计与蛋白质连接的肽时,主流方法依赖于两种途径:一是通过折叠优先范式生成序列,其中序列或结构作为轨迹进行迭代改进;二是通过从随机序列空间进行结构推导。
问题在于:穷举序列搜索和轨迹计算的规模与 20^n 成正比。结构优先方法可以规避这个问题,但许多方法需要反向折叠来恢复序列——这一步骤本身并不能考虑结合能。
LigandForge 则无需这些步骤,且不影响序列质量。
作者提出:如果我们彻底颠覆这种范式会怎样?与其随机生成序列或试图将结构当作锁和钥匙来匹配,不如构建一种新的方法,学习和理解相互作用的物理机制,而不仅仅是氨基酸和结构空间,那会怎样?
最初,作者从晶体数据中已知的蛋白质-蛋白质相互作用中提取并设计了360,000个突变肽,并用多层热力学数据对其进行注释,不仅提供了拓扑结构和坐标图,还提供了能量和结合图。
然后,训练了一个离散扩散、热力学监督的神经网络——LigandForge——以基于潜在蛋白质界面口袋特征同时生成氨基酸标记和连续能量输出。
这是一个参数量约为2500万、速度极快、质量极高的模型。
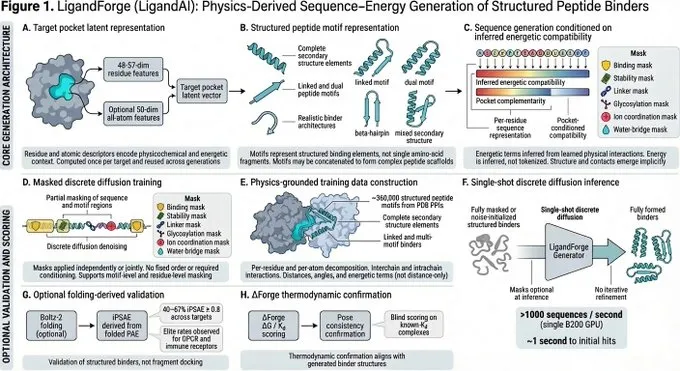
LigandForge 将氢键能量、盐桥和吉布斯自由能直接整合到训练损失中。
该模型不仅知道残基应该位于某个位置,还知道它为什么应该位于该位置。
与 Bindcraft 和 Boltzgen 直接比较时,作者的分层监督离散扩散神经网络在界面质量(ipSAE、pTM、iPTM、pLDDT)和计算结合能(∆G、Kd)方面达到或超过了最先进的基准。
DeltaForge是热力学评分引擎,根据实验结合数据进行校准(在 PPB-Affinity 肽基准测试中 r = 0.83),在 116 个评分目标中,有 85 个(73%)预测结合物低于 100 nM,62 个(53%)低于 10 nM,35 个(30%)低于 1 nM。
在针对历史上肽生成失败的靶点(TNF-α、PD-L1、VEGF-A、IL-7Rα、HER2)的五靶点基准测试中,LigandForge 在 3.4 分钟内生成了 150,000 个候选分子,并针对所有五个靶点生成了预测的低于 100 nM 的结合物(从 576 个折叠结构中总共生成了 23 个)。
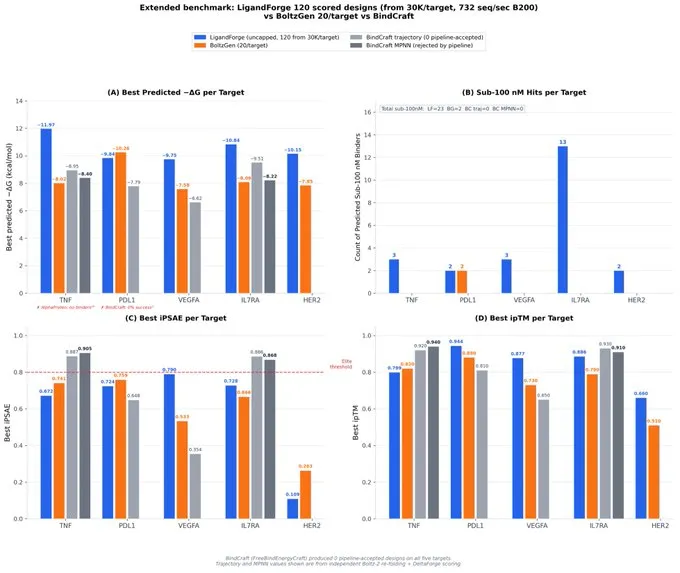
对于同一组靶点,与已发表的文献一致,BindCraft 完全未能生成任何可接受的设计,而 BoltzGen 在 100 个设计中仅对一个目标获得了 2 个命中。
此外,当使用 Boltz-2 对 16,475 个肽进行折叠以进行验证和进一步的热力学评分时,LigandForge 能够生成正构口袋嵌入肽,用于血清素和多巴胺受体,而肽结合在进化上没有任何先例。
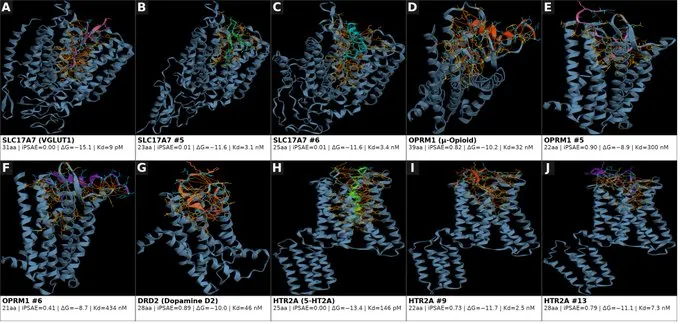
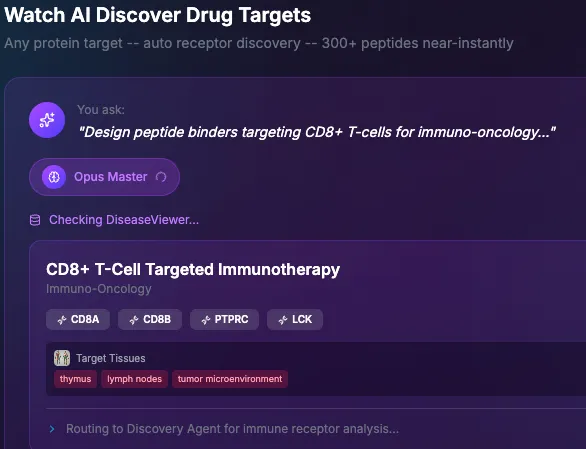
LigandForge可直接处理受体多聚体,无需预处理或定义热点区域。作者对 CD8A–CD8B 和 CD3D–CD3E 的异二聚体复合物以及具有空位配对的 KIT-SCF 四聚体进行了基准测试。只需提供受体或蛋白质靶标,即可获得结合物。
这仅仅是 LigandForge 的开始,它不仅仅是结合剂——它能够大规模进行推断,几乎可以瞬间针对任何靶点生成细胞和组织特异性肽。
立即注册参与作者的公开测试版,即可设计针对您靶点的肽:https://ligandai.com/
 请备注:姓名_研究方向_公司/学校
请备注:姓名_研究方向_公司/学校