 夜雨聆风
夜雨聆风





GEO 实战避坑系列①:GEO 下载的数据到底该用哪个文件?
前面我们聊了不少转录组分析里的基础问题。比如差异分析和富集分析有什么区别,DESeq2、limma、edgeR 怎么选,logFC、P 值、FDR 阈值怎么定,火山图、热图、PCA 到底怎么看。
这些内容更多是在讲“方法怎么理解”。但真到自己开始做 GEO 数据时,很多人会发现,最先卡住的往往不是差异分析,也不是富集分析,而是一个特别基础的问题:
GEO 页面里这么多文件,到底该下载哪个?这个问题看着简单,实际很容易出错。
很多人打开一个 GSE 数据集,看到页面上有一堆东西:
GSEGSMGPLSeries MatrixSOFTSupplementary filesRAW.tarcountFPKMTPMSRA Run Selector
看起来都像数据,但它们不是一回事。
有的是表达矩阵。有的是样本信息。有的是平台注释。有的是作者上传的补充文件。有的只是 GEO 整理出来的文本记录。
如果第一步文件就拿错了,后面差异分析、富集分析、WGCNA、免疫浸润、机器学习都会跟着偏。
所以从这篇开始,我准备写一个“GEO 实战避坑系列”。希望大家看了可以有所收获。
第一篇先讲最容易踩坑的地方:GEO 下载的数据,到底该用哪个文件?
一、先别急着下载,先看懂 GEO 页面
很多人打开 GEO 页面后,第一反应就是找下载按钮。但更稳的做法是,先把页面上的几个区域看懂。
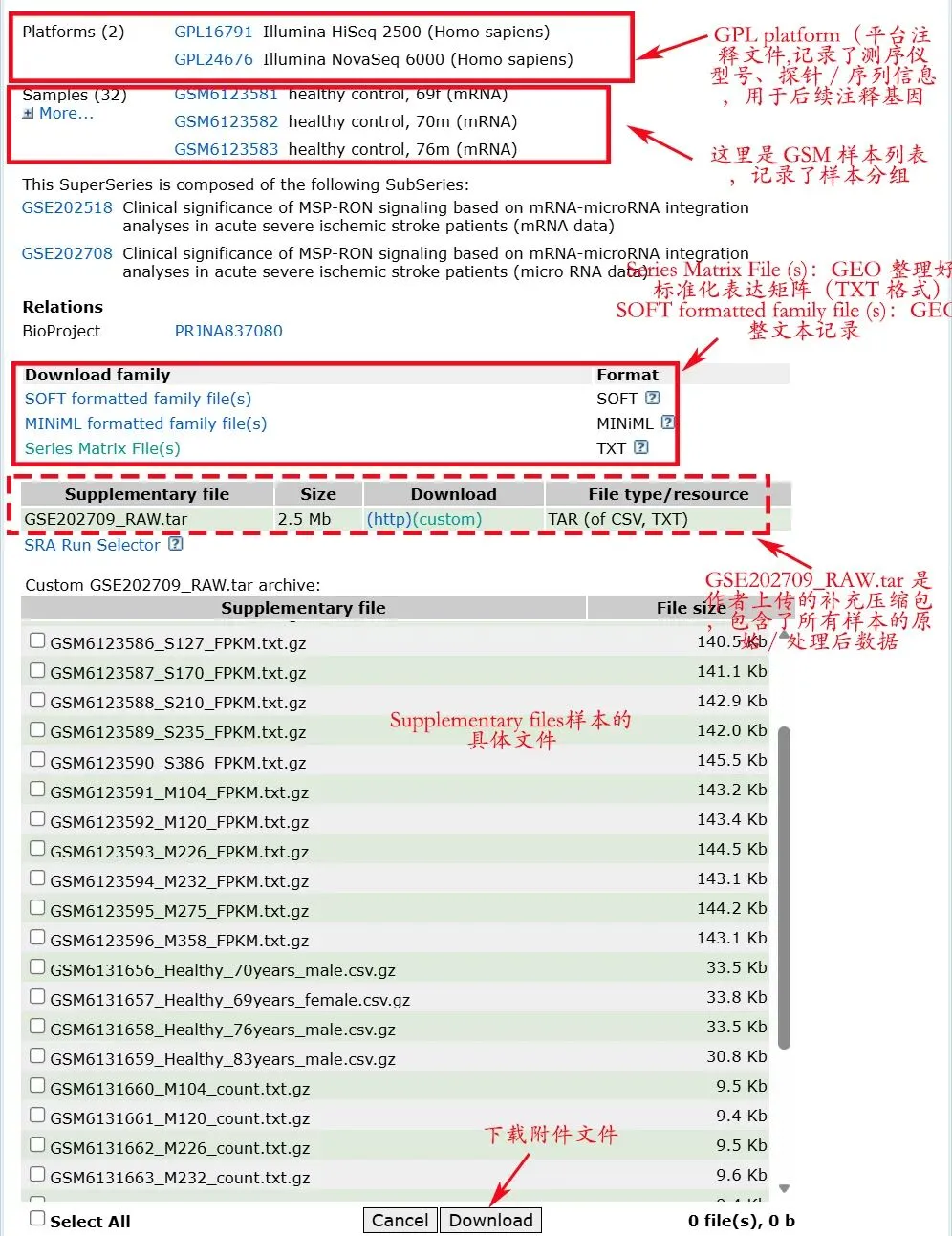
这张图里,其实已经把 GEO 页面最常见的几个区域都标出来了。最上面是 Platforms,也就是平台信息。
比如图中有 GPL16791 和 GPL24676,分别对应 Illumina HiSeq 2500 和 Illumina NovaSeq 6000。这里的 GPL 不是表达矩阵。
它记录的是这个数据用的什么平台、什么测序仪、什么芯片,或者平台相关的注释信息。对于芯片数据来说,GPL 很重要,因为后面经常要靠它把 probe ID 注释成 gene symbol。
对于 RNA-seq 数据来说,GPL 更多是记录测序平台和实验来源,不一定直接用于差异分析。
再往下是 Samples,也就是 GSM 样本列表。每一个 GSM 通常对应一个样本。比如图里可以看到 healthy control、不同年龄、不同性别等信息。但要注意,GSM 只是样本编号,不等于分组表已经整理好了。
真正做差异分析之前,你还要把每个 GSM 对应到具体分组。
比如:
control 还是 disease;normal 还是 tumor;mild 还是 severe;treated 还是 untreated。
这些信息有时写在 title 里,有时写在 characteristics 里,有时藏在作者上传的 sample information 表里。所以,看到 GSM 列表,只能说明“有哪些样本”,不能说明“分组已经能直接用了”。
二、Download family:这里有 GEO 整理好的文件
图中间的 Download family 区域,一般会看到这几类文件:
SOFT formatted family fileMINiML formatted family fileSeries Matrix File(s)
其中,很多人最常用的是 Series Matrix File(s)。它通常是 GEO 整理好的表达矩阵和样本信息,尤其在芯片数据里很常见。很多 GEOquery 下载下来的数据,本质上就是这个 series matrix。
它的优点是方便。下载快,读取也方便,通常可以直接拿到表达矩阵和样本注释。
但它也有几个问题。
第一,它不一定是原始表达值。
有些 series matrix 已经做过标准化。有些已经 log 转换。有些做过背景校正。有些甚至经过作者自己的过滤。
第二,它经常还是 probe ID。
尤其是芯片数据,第一列可能是:204016_at,213746_s_at,ILMN_1762337这些不是 gene symbol,而是探针编号。如果直接把 probe 当基因名用,后面分析就会乱。
第三,series matrix 里的样本信息不一定干净。
分组信息可能藏在 title、source_name、characteristics_ch1 这些字段里,而且命名不一定统一。所以 series matrix 可以用,但不能闭着眼用。至少要先确认三件事:
表达值是什么尺度;第一列是 probe、symbol 还是 Ensembl;样本分组到底在哪一列。
三、Supplementary files:很多时候这里才是重点
GEO 页面里最容易被忽略,但实际最重要的,往往是 Supplementary files。
图里可以看到一个文件:GSE202709_RAW.tar
很多人一看到 RAW.tar,就以为这是 fastq 原始测序数据,需要重新比对。
其实不一定。
很多 GEO 的 RAW.tar 只是一个压缩包,里面装的是作者上传的补充文件,可能是 txt、csv、count、FPKM、TPM 等处理后的结果。
比如图中点开以后,可以看到很多具体文件:
GSM6123586_S127_FPKM.txt.gzGSM6123591_M104_FPKM.txt.gzGSM6131660_M104_count.txt.gz
这就说明作者上传的不只是一个笼统的 RAW 包,而是每个样本对应的表达文件。
有的是 FPKM。有的是 count。有的是 csv。有的是 txt。
这类文件往往比单纯的 series matrix 更值得仔细看。
尤其是 RNA-seq 数据。很多 RNA-seq 的 series matrix 并不一定适合直接做分析,反而 supplementary files 里会有作者整理好的 count matrix、FPKM 或 TPM 文件。
所以拿到一个 GEO 数据集,不要只下载 Series Matrix。一定要看 supplementary files 里面到底有什么。
如果里面有 raw count,那做 DESeq2、edgeR 会更合适。 如果里面有 FPKM 或 TPM,那更适合做表达展示、相关分析、免疫浸润、GSVA、机器学习等。 如果里面有 sample annotation 或 clinical information,那它可能就是整理分组和临床信息的关键文件。
很多 GEO 分析不是输在代码,而是输在这里:还没搞清楚下载的是矩阵、注释、样本表,还是补充文件,就已经开始跑差异分析了。
四、GSE、GSM、GPL,先把这三个编号分清楚
GEO 页面里最常见的三个编号是 GSE、GSM、GPL。
它们很容易混,但含义不一样。
|
|
|
|
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
比如一个 GSE 里可以有几十个 GSM 样本。
这些 GSM 可能来自不同分组,比如正常、疾病、治疗前、治疗后。同一个 GSE 也可能包含多个 GPL 平台。比如图中就有两个平台,一个是 HiSeq 2500,一个是 NovaSeq 6000。
这时候就要小心。如果一个 GSE 里包含多个平台,不能直接把所有样本混在一起分析。
你要先确认:
这些平台对应的是不是同一种组学数据;是不是一个是 mRNA,一个是 miRNA;是不是不同测序批次;表达矩阵能不能合并;是否需要分开分析。
图里这个数据集还提示它是一个 SuperSeries,下面包含 SubSeries。
有些 GSE 是总合集,下面还分 mRNA 数据、miRNA 数据、甲基化数据、单细胞数据等。
如果你要做 mRNA 分析,就要找对应的 mRNA 子数据集,不要把 miRNA 或其他组学文件混进来。
五、count、FPKM、TPM,不能乱用
RNA-seq 里最常见的表达文件,大概有这几种:raw count、FPKM、TPM、log2(TPM+1)它们不能混着用。
|
|
|
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
如果你要做 DESeq2 或 edgeR,最好用 raw count。不要把 TPM、FPKM 当成 count 塞进去跑 DESeq2。
这是非常常见的错误。
因为 DESeq2 和 edgeR 是基于计数数据建模的,TPM、FPKM 已经经过了长度和测序深度校正,不再是原始计数。
如果你手里只有 FPKM 或 TPM,也不是完全不能分析。可以考虑 limma,或者用于 GSVA、ssGSEA、免疫浸润、相关性分析、机器学习等。
关键是方法要和数据类型匹配。文件拿错了,后面结果再漂亮也不稳。
六、芯片数据和 RNA-seq 数据,选文件思路不一样
如果是芯片数据,经常会用 series matrix。
流程是:
下载 series matrix;读取表达矩阵;下载或读取 GPL 平台注释;把 probe ID 转成 gene symbol;处理多个 probe 对应同一个 gene 的情况;再做差异分析。
这里最关键的是 probe 注释。芯片数据第一列常常不是基因名,而是探针。
一个 gene 可能对应多个 probe,一个 probe 也可能对应多个 gene。这一步如果处理不好,后面差异分析、WGCNA、机器学习都会受影响。如果是 RNA-seq 数据,更建议先看 supplementary files。因为作者很可能上传了 count、FPKM、TPM 这类文件。
这时你要根据分析目的选择:
做 DESeq2/edgeR,用 count;做表达展示和免疫分析,用 TPM 或 FPKM;做机器学习和相关分析,可以用 log 转换后的表达矩阵;做多队列整合,先统一 gene ID 和表达尺度。
所以,不同数据类型不能用同一个套路。
芯片先看 probe 和 GPL。RNA-seq 先看 count、FPKM、TPM。单细胞另说,要看 matrix、barcodes、features 或 h5 文件。
七、到底优先用哪个文件?
实际做 GEO 数据时,我一般建议按这个顺序看。
第一,先看 supplementary files。
尤其是 RNA-seq 数据。如果作者已经上传了 count matrix、TPM matrix、FPKM matrix 或样本注释文件,这些往往是最值得优先检查的。
第二,再看 series matrix。
如果是芯片数据,series matrix 很常用,但要结合 GPL 注释文件处理 probe。如果是 RNA-seq,也可以看,但不要默认它一定适合差异分析。
第三,必要时再用 RAW data 或 SRA。
如果没有现成表达矩阵,或者你想统一处理多个队列,才考虑从原始数据重新处理。这一步工作量大很多,不是每个项目都必须做。
第四,GPL 主要用于注释。
不要把 GPL 当表达矩阵。它是平台说明和注释信息,不是样本表达量。
总结一下:RNA-seq 优先看 supplementary files;芯片数据重点看 series matrix + GPL;找不到合适矩阵,再考虑 RAW/SRA;不要把平台文件当表达矩阵用。
八、拿到文件后,先做几个检查
不要刚下载完就差异分析。先检查这几件事。
第一,表达矩阵第一列是什么?
是 gene symbol?是 Ensembl?是 EntrezID?还是 probe ID?这个决定后面要不要注释转换。
第二,表达值是什么尺度?
是 raw count?是 FPKM?是 TPM?是 log2 转换后的值?还是芯片标准化表达值?这个决定你能用什么差异分析方法。
第三,样本名能不能对上?
表达矩阵里的样本名,必须和分组表里的样本名一一对应。顺序也要对上。这是最隐蔽的坑之一。代码不会报错,但结果可能全错。
第四,分组信息是否明确?
不要只看 title 里写了 control 或 disease 就直接用。最好整理出一个明确的 metadata 表:
samplegroupbatchplatformclinical information
这样后面分析才稳。
第五,基因 ID 有没有重复?
芯片 probe 转 gene 后经常重复。Ensembl 转 symbol 后也可能重复。重复基因要合并,否则很多下游分析都会出问题。
九、几个最常见的 GEO 文件错误
第一个,把 GPL 当表达矩阵。
GPL 是平台注释,不是表达数据。
第二个,把 probe 当 gene symbol。
芯片数据很常见,必须先注释。
第三个,把 FPKM/TPM 当 raw count 跑 DESeq2。
这个不推荐。
第四个,只下载 series matrix,不看 supplementary files。
很多作者真正有用的表达矩阵都放在补充文件里。
第五个,没整理 GSM 分组。
GSM 是样本编号,不等于分组表。
第六个,SuperSeries 和 SubSeries 没分清。
一个总 GSE 下面可能包含 mRNA、miRNA 或其他组学,不要混着用。
第七个,多平台样本直接合并。
不同 GPL、不同平台的数据,必须先确认能不能合并。
第八个,样本顺序没核对。
表达矩阵和分组表顺序错位,是非常隐蔽但很致命的问题。
十、最后说几句
GEO 实战的第一步,不是跑差异分析,也不是画火山图。而是先看清楚:
这个页面里每个文件到底是什么。
GSE 是数据集。GSM 是样本。GPL 是平台。Series Matrix 是 GEO 整理好的矩阵和样本信息。Supplementary files 是作者上传的补充文件,很多时候最关键。RAW.tar 不一定是 fastq,可能只是装着 count、FPKM、TPM 的压缩包。
count、FPKM、TPM 各有用途,不能乱塞进同一个方法。很多 GEO 分析翻车,不是因为代码不会写,而是从第一步就拿错了文件。文件拿错,后面分析跑得越顺,错得越远。所以做 GEO 数据,别急。
先看页面,再看文件,再选方法。
GEO 样本信息为什么对不上?表达矩阵、临床信息和分组表到底该怎么合并?