MS-MINT:专为大队列设计的 LC-MS 数据分析软件,3334个样本仅需25分钟!
|
|
MS-MINT: An Open-Source Data Analysis Software for Large-Scale Metabolomics Studies
|
|
期刊
|
Analytical Chemistry(分析化学顶刊)
|
|
作者团队
|
加拿大卡尔加里大学阿尔伯塔高级诊断中心 Ian A. Lewis 课题组
|
|
本文约
|
3600 字,预计阅读需要 8 分钟
|
一、传统代谢组学软件的四大致命缺陷
在介绍MS-MINT之前,我们必须先搞清楚:为什么现有的软件处理不了大规模数据?
论文一针见血地指出了传统工具(如XCMS、MZmine、EL-MAVEN等)的四个核心缺陷,每一个都直击痛点:
1. 内存依赖导致的批次误差
传统软件需要将整个数据集全部加载到内存中才能运行。当样本数超过1000个时,普通电脑的内存就会被耗尽,只能将数据拆分成多个批次分别处理。
但代谢组学分析天生具有非确定性:峰的选择、信号平均、峰排除等决策高度依赖当前批次的数据上下文。这就导致同一个代谢物在不同批次中可能得到完全不同的定量结果,引入无法消除的系统性误差。
2. 数据依赖的峰拟合算法
传统软件的自动峰拟合算法性能,完全取决于内存中加载的数据量和数据分布。分批处理时,算法会对每个批次单独优化参数,最终导致全队列的峰提取标准不一致。
3. 共洗脱代谢物区分困难
区分保留时间相近的共洗脱代谢物,一直是LC-MS分析的难题。在小规模研究中,我们可以通过人工目视检查逐个修正,但在包含数万甚至数十万信号的大规模数据集中,这完全是不可能完成的任务。
更糟糕的是,色谱柱性能会随着进样次数增加而逐渐漂移,进一步加剧了共洗脱峰的区分难度。
4. 分析流程无法记录与重复
传统代谢组学分析依赖大量的逐峰手动决策,而这些决策根本无法被精确记录。再加上算法本身的数据依赖性,导致几乎没有任何一项代谢组学研究能够被他人完全重复。
这四个问题相互交织,使得大规模代谢组学分析变成了一个耗时、费力、且结果不可靠的”黑箱”。
二、MS-MINT:基于ROI的革命性解决方案
针对上述所有问题,Lewis课题组提出了一个全新的分析范式:基于感兴趣区域(ROI)的确定性分析方法。
核心设计理念
MS-MINT没有沿用传统的”先找峰、再对齐、后定量”的思路,而是借鉴了NMR软件的设计,将每个代谢物定义为一个固定的保留时间+m/z窗口(ROI)。
软件不需要加载整个数据集,只需要从每个样本中提取这个ROI窗口内的数据进行分析。这一简单的改变,从根本上解决了传统软件的所有核心缺陷:
技术栈:全Python生态,开箱即用
MS-MINT完全基于Python生态系统开发,确保了跨平台兼容性和可扩展性:
-
-
GUI框架:Plotly Dash(网页式界面,无需复杂安装)
-
数据读取:pigyxml(高效解析mzML/mzXML标准格式)
-
数据存储:Apache Parquet(Snappy压缩)+ DuckDB(列式数据库)
-
-
软件可通过PyPI一键安装:pip install ms-mint-app2,也可从课题组官网免费下载。
三、五步法标准化工作流
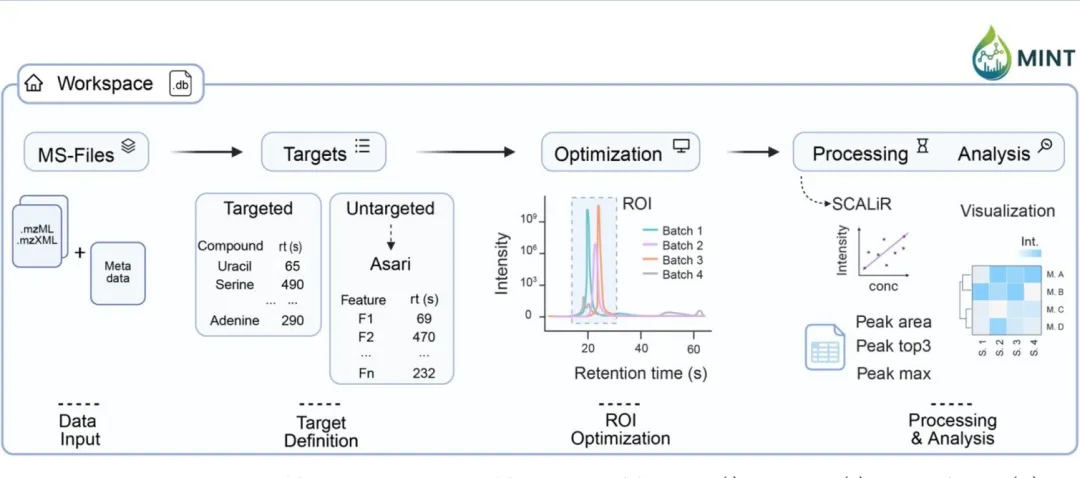
MS-MINT将整个代谢组学分析流程标准化为五个独立模块,所有步骤和结果都封装在一个可移植的”Workspace(工作空间)”中,确保完全的可重复性。
【图1】MS-MINT处理工作流:整个流程分为数据输入、目标定义、ROI优化、数据处理和结果分析五个步骤,所有输出都保存在可移植的工作空间和数据库中。
1. 数据输入:一键批量导入
-
支持批量导入mzML/mzXML格式的LC-MS文件(兼容所有主流仪器厂商)
-
自动将原始数据转换为Parquet格式(体积缩小50%-80%,读取速度提升10-100倍)
-
转换后的数据存入DuckDB数据库,支持超内存数据集的并行查询
-
可导入包含批次信息、样本类型、分组信息等的元数据表格
2. 目标定义:支持靶向+非靶向双模式
-
靶向分析:导入预定义的目标表,包含代谢物名称、保留时间和m/z值
-
非靶向分析:集成Asari代谢组学工具,自动检测所有特征并生成初始目标列表
-
3. ROI优化:可视化精确调整
-
图形化显示提取离子色谱图(XIC),高亮标注ROI区域
-
支持鼠标拖拽直接调整ROI边界,所有修改自动保存到数据库
-
局部保留时间对齐:针对色谱漂移问题,可基于参考峰进行逐样本保留时间校正
-
4. 数据处理:多指标定量+绝对定量
-
-
计算多种定量指标:峰面积(梯形法)、峰面积top3、最大强度保留时间等
-
可选指数修正高斯(EMG)分布拟合,对复杂峰形进行更精确的计算
-
集成SCALiR工具,利用标准曲线自动计算代谢物的绝对浓度
5. 结果分析:集成式统计可视化
-
-
提供PCA、t-SNE、小提琴图、热图、聚类分析等多种统计可视化工具
-
支持z-score归一化、log2转换、Rocke-Durbin转换等常用预处理方法
-
四、硬核性能验证:3334个样本的实战测试
为了验证MS-MINT的性能,研究人员使用了一个包含3334个LC-MS谱图的大规模真实数据集。这个数据集专门为评估色谱性能漂移设计,包含超过3000次混合样本重复注射和192次标准品注射,是软件性能对比的黄金基准。
研究人员将MS-MINT与两款行业标杆软件进行了头对头对比:
-
EL-MAVEN:最流行的开源LC-MS数据处理引擎,以定量准确著称
-
peakPantheR:基于ROI的R包,专为大规模靶向分析设计
【图3】MS-MINT与主流软件的性能对比:(a) 与EL-MAVEN的定量一致性;(b) 与peakPantheR的处理速度对比;(c) 与peakPantheR的定量一致性。
1. 定量准确性:与专家手动分析几乎完全一致
首先对比的是MS-MINT与EL-MAVEN的定量结果。EL-MAVEN的分析由经验丰富的代谢组学专家手动完成,耗时数天。
结果令人震惊:MS-MINT的peak_area_top3指标与EL-MAVEN的PeakAreaTop指标的Pearson相关系数高达0.999!
仅在低强度峰区域存在轻微差异,这是由于低信噪比下不同软件的噪声处理策略不同导致的,属于正常现象。这证明MS-MINT的全自动算法,能够达到与专家手动分析相当的定量准确性。
2. 处理速度:25分钟 vs 数天
同样的3334个样本,MS-MINT在一台配备AMD Ryzen 7 2700X八核处理器和32GB内存的普通台式机上,仅用25分钟就完成了全部分析。
而同样的工作,专家用EL-MAVEN手动完成需要数天时间。
3. 与同类型ROI工具的对比
研究人员还将MS-MINT与同样基于ROI的peakPantheR进行了对比。结果显示:
-
定量一致性:Pearson相关系数r=0.970,结果高度一致
-
处理速度:MS-MINT平均比peakPantheR快约6倍
-
并行扩展性:随着CPU线程数增加,MS-MINT的性能提升更为显著。当使用16线程时,MS-MINT仅需约10分钟就能完成全部分析,而peakPantheR需要约60分钟。
五、MS-MINT的四大核心优势
1. 真正的超大规模数据处理能力
基于DuckDB的列式数据库架构,MS-MINT支持处理大于系统内存的数据集,无需分批处理,从根本上消除了批次误差。
2. 100%可重复的分析流程
所有分析参数(包括ROI定义、保留时间偏移、峰拟合方法、归一化方式等)都精确记录在数据库中,可一键导出为CSV文件。他人只需导入该文件,就能完全重复整个分析流程。
3. 集成式迭代分析工作流
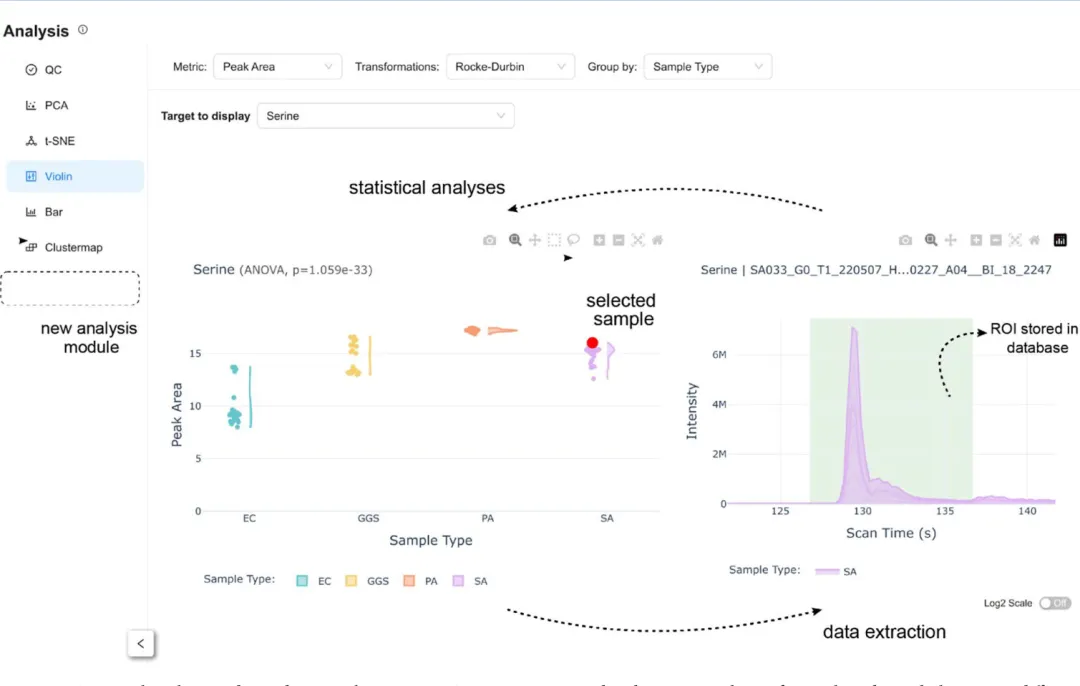
MS-MINT最具特色的功能是”统计-原始数据联动”。当你在统计可视化中发现异常值时,只需点击该样本点,就能立即查看对应的原始色谱图,并快速返回ROI优化步骤进行修正。
这一功能将传统软件中”处理→分析→发现问题→重新处理”的漫长循环,缩短为几秒钟的操作。
4. 开源免费,社区驱动
MS-MINT完全开源免费,采用模块化设计,允许社区开发者贡献新的功能和分析模块。这意味着它会随着代谢组学领域的发展而不断进化。
六、总结与展望
MS-MINT的出现,标志着大规模代谢组学分析进入了一个新的时代。它通过基于ROI的确定性分析方法,成功解决了传统软件面临的内存限制、批次误差、结果不一致和难以重复等核心问题。
对于科研人员来说,MS-MINT带来的改变是革命性的:
-
原本需要数周的大规模数据分析,现在只需几十分钟就能完成
-
-
科研人员可以将更多精力放在生物学问题的解读上,而不是繁琐的数据处理中
-
目前仅正式验证了Thermo Fisher仪器产生的数据,未来版本将扩展支持其他主流厂商
-
-
软件下载地址:https://www.lewisresearchgroup.org/software
写在最后
代谢组学作为连接基因组和表型组的桥梁,正在生命科学和医学研究中发挥着越来越重要的作用。但长期以来,数据分析能力的不足,严重制约了代谢组学技术的应用和发展。
MS-MINT这样的开源工具的出现,不仅解决了一个具体的技术问题,更降低了大规模代谢组学研究的门槛,让更多实验室能够开展大队列研究。我们相信,随着这类工具的不断完善,代谢组学必将在未来带来更多突破性的发现。
如果觉得这篇文章对你有帮助,欢迎点赞、在看、转发给更多需要的朋友。关注我们,获取更多代谢组学领域的前沿技术和工具解读。
 夜雨聆风
夜雨聆风