 夜雨聆风
夜雨聆风
OpenClaw Medical Skills正是瞄准这一痛点,将生物信息学的专业知识、标准化流程与工具能力,封装为可直接调用、可自动化执行的独立技能模块,彻底解决通用大模型在生信领域“只会说、不会做”“流程不闭环、结果不可靠”的核心问题。它不仅能连接GEO、TCGA、ENCODE等权威数据库,自动完成数据下载与预处理,还能无缝调用DESeq2、limma、Seurat等主流分析工具,实现从原始测序数据到科研图表的全流程自动化,更能将复杂的生信逻辑转化为自然语言指令,大幅降低生信分析的技术门槛。
由于篇幅有限(也考虑读者阅读情绪),本文作为续篇一,将聚焦生物信息分析的基础环节——《质控与读长处理》,详解其功能与应用价值,为后续生信分析筑牢基础。软件表格整理放于文末。
1
质控与读长处理
任何基因组学流程的首要关键关卡,是确保原始测序读长(reads)在进入下游分析前符合质量标准。此技能套件提供了测序质量控制与读长处理的端到端覆盖——从初始的 FASTQ 质量评估和接头修剪,到污染筛查、UMI 去重、比对,以及专门的 RNA-seq 质控。该合集同时支持短读长(Illumina)和长读长(Oxford Nanopore、PacBio)平台,为生产级流程提供 CLI 驱动的工具,并为程序化质量检查提供 Python(Biopython)实用程序。这 20 多项技能共同构成了基础层,变异检测、差异表达以及所有下游生物信息学分析均依赖于该基础层。
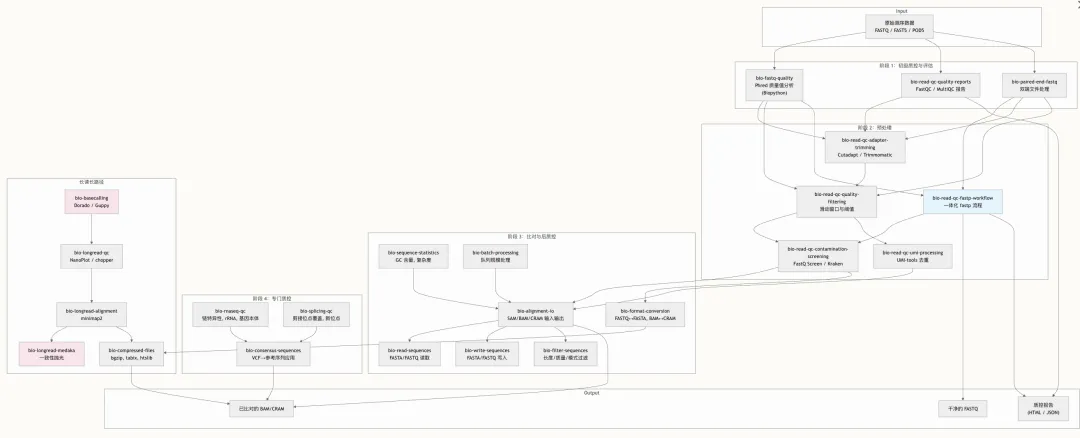
短读长处理流程
1.1 FASTQ 质量评估
bio-fastq-quality 使用 Biopython 的 Bio.SeqIO 提供对 Phred 质量值的程序化访问。它无需运行外部 CLI 工具,即可实现 Python 原生的质量值分析——计算单条读长平均值、按平均质量过滤、使用滑动窗口修剪低质量的 3' 端,并生成逐位置的质量分布图。当你需要将质控逻辑集成到自定义的 Python 流程中,而不是依赖 FastQC 的静态报告时,这项技能尤为实用。它还能处理 FASTQ 格式变体转换(Sanger/Illumina 1.3+/Solexa)以及质量编码的自动检测。
bio-read-qc-quality-reports 提供了标准化的报告层,使用 FastQC 进行单样本检查,并使用 MultiQC 进行队列级别的聚合。FastQC 生成包含逐碱基质量、接头含量、GC 分布、过度代表序列和重复水平等模块的 HTML 报告。MultiQC 将所有样本的这些结果聚合到一个交互式报告中。该技能涵盖从 FastQC 的 ZIP 输出中程序化提取数据、配置自定义接头序列和警告阈值,以及建立用于记录预处理改进效果的前后对比工作流。
1.2 接头修建
bio-read-qc-adapter-trimming 解决使用 Cutadapt(精准、灵活的模式匹配)和 Trimmomatic(针对双端通量优化,附带 Illumina 接头文件)去除测序接头污染的问题。Cutadapt 可处理单端、双端和链接接头场景,并对错误容忍度、最小重叠要求和操作模式(修剪、掩码、丢弃)进行细粒度控制。Trimmomatic 内置了 Illumina TruSeq、Nextera 和小 RNA 接头序列。该技能包含一份按平台划分的常见接头序列参考表,并提供了通过修剪后 FastQC 报告验证修剪效果的指导。
1.3 质量过滤
bio-read-qc-quality-filtering 实现了接头去除后的核心读长过滤逻辑。它涵盖了三种互补的方法:Trimmomatic 滑动窗口修剪(SLIDINGWINDOW:4:20 会移除在 4 个碱基上平均质量低于 20 的窗口)、带有逐读长和逐碱基阈值的 fastp 质量过滤,以及基于质量的 Cutadapt 修剪。该技能提供了开箱即用的 Trimmomatic“配方”——针对标准 RNA-seq、WGS 和扩增子测序等常见场景预配置的操作链。它还涉及了 NovaSeq/NextSeq 的 poly-G 修剪,这对于将无碱基调用(N)编码为 G 的平台来说是必不可少的。
1.4 一体化:fastp 工作流
bio-read-qc-fastp-workflow 是大多数 Illumina 预处理任务的推荐起点。fastp 将接头修剪、质量过滤、质量修剪、poly-X 修剪、N 碱基处理、去重、碱基校正(双端)、读长合并和 UMI 处理整合到一个单遍处理的工具中,并内置了 HTML 和 JSON 报告。该技能提供了三个完整的工作流模板:


fastp 与独立工具对比:在大多数用例中,bio-read-qc-fastp-workflow 用单条命令替代了 bio-read-qc-adapter-trimming → bio-read-qc-quality-filtering → bio-read-qc-quality-reports 这三步链条。仅当你需要 Cutadapt 的高级模式匹配(例如,带有特定锚点的链接接头)或 Trimmomatic 的传统 Illumina 接头处理时,才应使用独立工具。
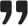
1.5 污染筛查
bio-read-qc-contamination-screening 使用 FastQ Screen(基于比对)或 Kraken(基于 k-mer)检测跨物种污染。FastQ Screen 将读长与多个参考数据库并行比对,并报告映射到每个数据库的比例,非常适合检测微生物样本中的人类污染,反之亦然。该技能涵盖使用预构建数据库进行配置文件设置、自定义 Bowtie2 索引创建、过滤代码解读(映射质量阈值),以及与 MultiQC 集成以生成队列级别的污染报告。预期的污染特征因样本类型而异,该技能提供了有关区分真实污染与预期的多重映射的解读指南。
1.6 UMI处理
bio-read-qc-umi-processing 处理完整的 UMI(唯一分子标识符)工作流,用于在纠错测序方案中进行准确的分子定量。使用 UMI-tools,它涵盖了从三种常见架构中提取 UMI:嵌在读长序列中的 UMI(使用 umi_tools extract --bc-pattern)、位于单独索引读长中的 UMI,以及具有可变长度条形码的复杂模式。去重步骤支持多种算法——定向(directional)、唯一(unique)和基于聚类(cluster-based)的算法,并附带基于实验设计的方法选择指南。该技能提供了 RNA-seq 的基因级去重和 CellRanger 后单细胞工作流作为完整示例。此外,它还记录了去重率的解读:低于 30% 的比率表明文库复杂度低,而高于 80% 的比率可能暗示过度扩增。
1.7 双端 FASTQ 处理
bio-paired-end-fastq 提供了基于 Biopython 的实用程序,用于处理双端读长文件这一常见但极易出错的繁琐任务。它涵盖了带验证的配对迭代(确保 R1/R2 读长通过头部匹配)、联合过滤(当任一读长未达到质量/长度阈值时丢弃两端读长)、内存高效的 gzip 压缩配对处理、交替排列与拆分交替排列,以及通过命名约定自动检测配对文件。该技能弥合了原生处理配对文件的 CLI 工具(fastp、Trimmomatic PE 模式)与需要程序化配对管理的自定义 Python 流程之间的鸿沟。
长读长处理流程
由于长读长测序(Oxford Nanopore 和 PacBio)在错误特征、文件格式和质量特性上存在根本差异,因此需要一条截然不同的处理路径。
1.8 碱基识别
bio-basecalling 涵盖了将原始电信号(POD5/FAST5)转化为核苷酸序列的过程。Dorado 是当前 Oxford Nanopore 数据推荐的碱基识别工具,取代了已弃用的 Guppy。该技能文档记录了跨 R10.4.1 和 R9.4.1 化学试剂的模型选择、GPU 配置、用于实现最高准确率的 duplex calling、修饰碱基检测(5mC、5hmC),以及在碱基识别过程中的 demultiplexing。数据管理部分包含了 POD5 文件处理实用程序(从 FAST5 转换、合并、检查、子集提取)。一个完整的流程示例串联了碱基识别 → 质量过滤 → 比对 → 抛光。
1.9 长读长质量控制
bio-longread-qc 提供了专门针对长读长数据的质控层,在此类数据中,读长长度分布和 N50 是首要的质量指标,而非逐碱基的 Phred 质量值。NanoPlot 可生成读长长度与质量的散点图、N50 直方图和累积产量曲线。chopper 提供按读长和平均质量进行快速过滤的功能(例如,chopper -q 10 -l 1000 保留 Q≥10 且长度 ≥1kb 的读长)。NanoFilt 提供了另一种过滤路径,而 Porechop 用于处理 ONT 接头修剪。该技能还记录了如何使用 pycoQC 从测序汇总文件进行碱基识别级别的质量评估。
1.10 长读长比对
bio-longread-alignment 涵盖了针对三种主要长读长比对场景的 minimap2 配置:Oxford Nanopore 读长(-x map-ont)、PacBio HiFi 读长(-x map-hifi)以及 PacBio CLR 读长(-x map-pb)。其他预设包括用于长读长 RNA-seq 的剪接感知比对(-x splice 或用于短读长的 -x splice -uf -k14)以及组装序列到参考序列的映射(-x asm5/asm10/asm20)。该技能涵盖输出格式选择(BAM 与 PAF)、次要/补充比对的保留、多文件批处理、比对统计信息生成,以及用于下游分析的 PAF 到 BED 的转换。
1.11 一致性抛光
bio-longread-medaka 应用 Oxford Nanopore 的 Medaka 工具,利用训练好的神经网络进行一致性序列抛光和变异检测。模型的选择取决于所使用的碱基识别工具(Dorado 或 Guppy)及化学试剂版本,该技能提供了一份用于正确模型配对的查找表。它支持全基因组抛光、仅特定区域抛光、多轮迭代抛光,以及直接从现有 BAM 文件进行变异检测而无需重新比对。GPU 加速可显著提升大型基因组的处理速度。
序列输入输出与实用技能
多项实用技能通过处理文件格式操作、批处理和统计汇总,为质控和处理流程提供了底层支撑。
这些技能充当了专门处理阶段之间的连接纽带。例如,bio-batch-processing 能够跨数百个样本以队列规模执行任何质控工作流,而 bio-compressed-files 则通过 bgzip/tabix 索引确保大型比对文件的高效存储和随机访问查询。
专门 RNA-seq 质控
bio-rnaseq-qc 解决了 RNA 测序中超越通用 FASTQ 级别指标的特定质量问题。它涵盖了六项关键的 RNA-seq 特有评估:
rRNA 污染检测,使用 SortMeRNA 或针对 rRNA 数据库进行 BLAST——预期水平从 <5%(poly-A 筛选)到 >50%(总 RNA/去核糖体 RNA)不等
链特异性验证,使用 RSeQC infer_experiment 或 Salmon 的链特异性检测——对于正确的下游定量至关重要
基因本体覆盖度分析,使用 RSeQC geneBody_coverage——识别指示降解或建库方案问题的 3' 或 5' 偏倚
读长分布,跨基因组特征(外显子、内含子、基因间区),使用 RSeQC read_distribution
转录本完整性评分,用于单转录本质量评估
插入片段大小估计,针对双端文库使用 Picard CollectInsertSizeMetrics
该技能提供了一套完整的端到端 RNA-seq 质控流程,以及一个基于 Python 的质控汇总生成器,带有用于自动化评估的通过/失败阈值。与 MultiQC 的集成可生成一份将 RNA-seq 特定指标与标准 FastQC 输出相结合的统一报告。
bio-splicing-qc 将 RNA-seq 质量评估延伸至剪接领域,评估剪接位点读长覆盖度并识别新型剪接位点。它与 bio-splicing-quantification(PSI/包含水平计算)和 bio-sashimi-plots(剪接位点可视化)一起,形成了一个以剪接为重点的质控子流程,将通用读长质控与下一目录部分涵盖的差异表达和转录组学技能连接起来。
工具选择指南
选择合适的技能组合取决于你的测序平台、通量需求和下游分析需求。
安装与集成
上一篇粉丝有留言问如何用,如果大家需要用到这篇文章的功能,可以集成给你的小龙虾或者claue code。当然如果是内网环境的小龙虾,工具需要预先安装才能调用哦。
每项技能的 SKILL.md 前置元数据指定了 tool_type(cli 或 python)和 primary_tool,指示该技能提供的是命令行模式还是 Python API 用法。CLI 技能要求在 Agent 环境中安装相应的工具;Python 技能需要 Biopython 及任何指定的依赖包。版本兼容性记录在每个技能文件的顶部,在使用前应进行验证。
git clone https://github.com/MedClaw-Org/OpenClaw-Medical-Skills.git# 安装质控与读长处理技能QC_SKILLS=("bio-fastq-quality""bio-read-qc-adapter-trimming""bio-read-qc-quality-filtering""bio-read-qc-fastp-workflow""bio-read-qc-quality-reports""bio-read-qc-contamination-screening""bio-read-qc-umi-processing""bio-paired-end-fastq""bio-rnaseq-qc""bio-longread-qc""bio-longread-alignment""bio-basecalling""bio-longread-medaka""bio-alignment-io""bio-format-conversion""bio-batch-processing")for skill in "${QC_SKILLS[@]}"; docp -r OpenClaw-Medical-Skills/skills/$skill ~/.openclaw/skills/done
📌 往期回顾
第1期:开源医疗AI的“武器库”OpenClaw Medical Skills:869个技能,拿走不谢
第2期:OpenClaw Medical Skills 生信技能详解|质控与读长处理
📢 下期预告:《OpenClaw Medical Skills 生信技能详解|变异监测与注释》,敬请期待
附1 短读长处理流程软件汇总表:
附2长读长处理流程软件汇总表:
附3 专门 RNA-seq 质控软件汇总表:
