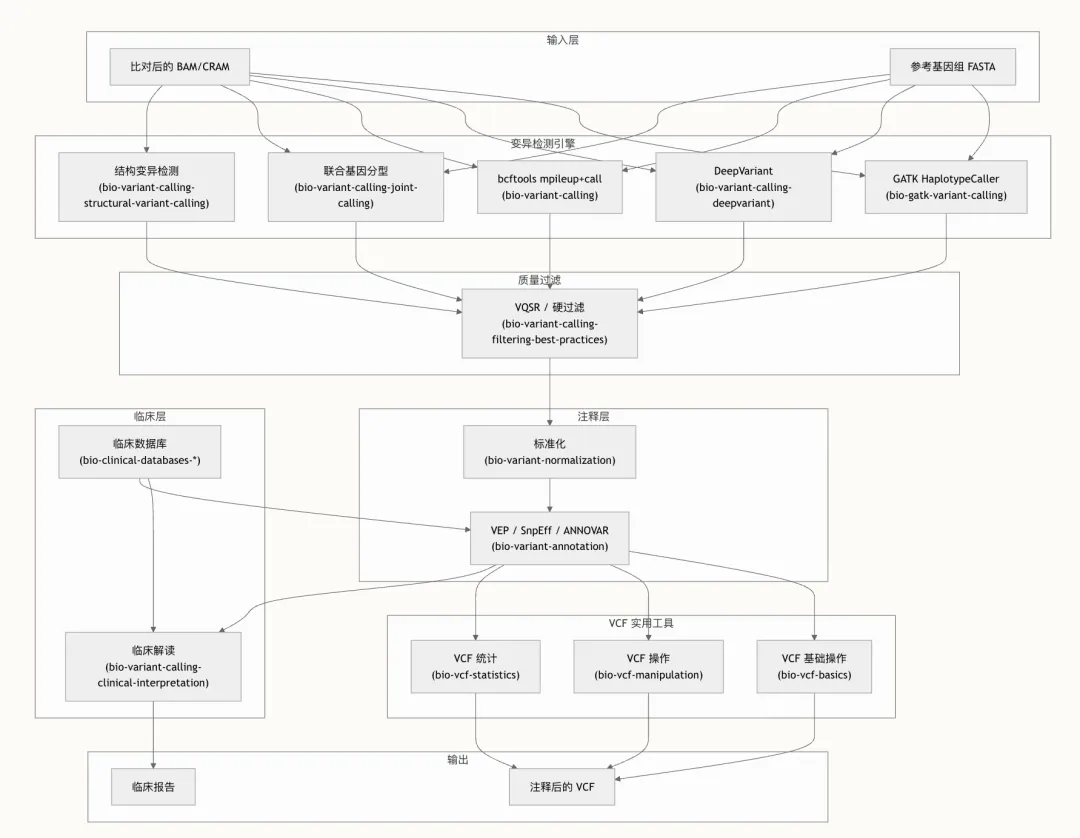
变异检测与注释构成了任何基因组学分析流水线的核心——将原始的比对reads转化为具有临床和功能可解释性的变异目录。在 OpenClaw Medical Skills 中,该领域由丰富的 bio-* skills 生态系统提供支持,覆盖了从生成 pileup 到临床分类的整个生命周期。本页提供了一份结构化指南,旨在帮助你理解、选择和组合这些技能,以构建生产级的变异分析工作流。本仓库中的变异检测与注释领域包含 17 个专用技能,按功能划分为四个层级:变异检测引擎、变异注释工具、VCF 操作实用程序以及临床解读层。这些技能由上游的质量控制能力OpenClaw Medical Skills 生信技能详解|质控与读长处理和下游分析(涵盖在精准医学与变异解读中)作为补充。
该架构遵循严格的线性依赖关系:比对后的 reads 必须先进行变异检测,然后进行质量过滤,接着进行标准化,最后进行注释——临床解读则消耗完全注释后的输出。根据分析的具体需求,每一层都可以独立替换。
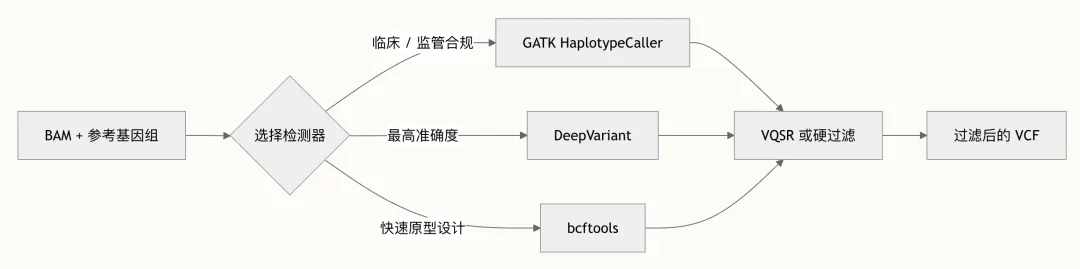
提供三种主要的变异检测,每种都针对不同的应用场景和性能特征进行了优化。选择正确的检测器是流水线架构中最具影响力的单项决策。
bio-variant-calling-deepvariant中等(WGS 约 4-6 小时,GPU 约 1-2 小时)2.1 GATK HaplotypeCaller — 临床标准(bio-gatk-variant-calling)
GATK HaplotypeCaller 依然是临床和研究领域胚系变异检测的黄金标准。bio-gatk-variant-calling 技能实现了完整的 GATK 最佳实践工作流,包括 BQSR 预处理、单样本及基于 GVCF 的队列检测、VQSR、硬过滤以及新兴的 CNN 评分过滤。建议所有队列分析均使用 GVCF 工作流,因为它能够跨样本进行联合基因分型,从而提高罕见变异的灵敏度。
完整的单样本流水线包含五个阶段:BaseRecalibrator → ApplyBQSR → HaplotypeCaller(GVCF 模式)→ GenotypeGVCFs → VariantFiltration。对于队列分析,在联合基因分型之前,需通过 GenomicsDBImport 合并各样本的 GVCF。当变异数量不足以进行 VQSR 时(通常为外显子组或小面板),则应用硬过滤阈值:QD > 2.0, FS < 60 (SNP) / < 200 (indel), MQ > 40, SOR < 3 (SNP) / < 10 (indel)。
2.2 DeepVariant — 深度学习级别的准确度(bio-variant-calling-deepvariant
DeepVariant 代表了一种范式转变,它将 read pileup 转换为图像张量,并使用卷积神经网络对其进行分类。bio-variant-calling-deepvariant 技能涵盖了基于 Docker 的部署、所有五种模型类型(WGS、WES、PACBIO、ONT_R104、HYBRID_PACBIO_ILLUMINA)、GPU 加速以及基于 GLnexus 的联合基因分型。DeepVariant 在 GIAB 基准测试中始终名列前茅,当准确度是首要约束条件时,它是理想的选择。
其一个关键优势是无需修改流水线即可原生支持长读长测序平台——只需切换 --model_type 参数即可。该技能还包含了使用 hap.py 针对 GIAB 真实数据集进行基准测试的指南,从而实现准确的定量评估。
2.3 bcftools — 轻量且快速(bio-variant-calling)
bio-variant-calling 技能提供了从 BAM 到 VCF 最快的路径,通过管道将 bcftools mpileup 传递给 bcftools call。虽然对于复杂变异,其准确度低于 GATK 或 DeepVariant,但它在快速原型设计、靶向区域分析以及计算资源有限的场景中表现优异。该技能涵盖了质量过滤(-q/-Q 阈值)、多样本检测、靶向区域限制(BED 文件)、按染色体并行化以及全面的注释标签配置(DP、AD、ADF、ADR、GQ、PL)。
如何在检测器之间做选择:对于临床级流水线,GATK HaplotypeCaller 仍然是最稳妥的选择,因为它拥有广泛的验证文献和监管机构的熟悉度。当基准测试证明的准确度至关重要(例如罕见病诊断)且计算预算允许时,应首选 DeepVariant。bcftools 最好保留用于探索性分析或作为验证交叉检查。
原始的变异检测结果包含假阳性,必须在注释前将其去除。两个互补的技能应对了这一关键的质量控制关卡。
3.1 VQSR 与硬过滤
bio-variant-calling-filtering-best-practices 技能涵盖了基于机器学习的 VQSR(变异质量分数校准)和固定阈值的硬过滤。VQSR 使用已知变异资源集构建高斯混合模型——HapMap(真实 SNP,先验 15.0)、Omni(训练 SNP,先验 12.0)、1000 Genomes(训练 SNP,先验 10.0)、Mills Indels(真实 indel,先验 12.0)以及 dbSNP(已知变异,先验 2.0)——以将变异分类为 PASS 或已过滤。硬过滤是针对数据集过小而无法支持 VQSR 时的备选方案,对关键注释应用基于阈值的表达式。
GATK 技能还记录了新兴的 CNNScoreVariants 深度学习过滤器作为 VQSR 的替代方案,它使用在参考序列上下文上训练的卷积神经网络对变异进行评分,并按 tranche 灵敏度进行过滤。
3.2 关键过滤注释
注释通过添加功能影响、人群频率、临床意义和致病性预测,将基础的 VCF 转化为描述丰富的变异目录。bio-variant-annotation 技能作为中央枢纽,涵盖了四个注释引擎:bcftools annotate/csq、Ensembl VEP、SnpEff 和 ANNOVAR。
4.1 注释工具对比
4.2 Ensembl VEP — 全能注释器
VEP(Variant Effect Predictor)是功能最丰富的选项,被指定为注释技能的主要工具。配合 --everything 标志运行,可在单次调用中启用 SIFT 预测、PolyPhen-2 评分、HGVS 命名法、基因符号、规范转录本选择、1000 Genomes 频率、gnomAD 频率以及 PubMed 参考文献。该技能记录了用于 CADD 评分、dbNSFP 预测器(SIFT、PolyPhen、REVEL 等 20 多种)以及 SpliceAI 剪接效应预测的插件集成 。
完整的注释流水线包含三个阶段:变异标准化(左对齐和分解)、使用 --everything --pick 进行 VEP 注释,以及基于影响程度对 HIGH/MODERATE 后果进行过滤。--pick 标志为每个变异选择一个代表性转录本,从而简化下游过滤。
4.3 SnpEff + SnpSift — 快速批量处理
SnpEff 使用本地基因组数据库提供快速的影响注释,将变异分为四个影响层级:HIGH(获得终止密码子、移码、剪接供体/受体)、MODERATE(错义、非移码插入缺失)、LOW(同义、剪接区域)和 MODIFIER(内含子、基因间、UTR)。SnpSift 通过数据库注释(dbSNP rsID、ClinVar 临床意义、dbNSFP 致病性评分)和基于表达量的过滤对其进行了扩展。多个 SnpSift 操作可在流水线中串联,以实现单次遍历的全面注释。
4.4 bcftools annotate — 轻量级字段操作
对于更简单的注释需求——例如从 dbSNP 添加 rsID 或附加基于 BED 的自定义区域标签——bcftools annotate 提供了最快的路径。它根据位置和等位基因与外部 VCF、BED 或 TAB 文件进行匹配,复制指定的 ID 或 INFO 列。配套的 bcftools csq 命令使用 GFF3 基因模型预测功能后果,而无需安装完整的注释引擎 。
4.5 致病性预测器与临床意义
> 20 (前 1%), > 30 (前 0.1%)ClinVar 临床意义分类遵循 ACMG/AMP 框架:Pathogenic(致病)、Likely_pathogenic(可能致病)、Uncertain_significance(意义不明,VUS)、Likely_benign(可能良性)和 Benign(良性)。注释技能提供了这些代码与其临床解读之间的映射关系,这些映射将直接输入到临床解读层。
4.6 VCF 操作与统计
在进行注释之前,变异通常需要标准化(左对齐和多等位基因分解)。在注释之后,VCF 文件需要进行合并、拆分、统计汇总和子集化。三个专用技能构成了 VCF 实用工具层。
bio-variant-normalization标准化是准确注释的前提:如果没有左对齐,相同的变异在不同检测器中可能会有不同的表示形式,从而导致遗漏数据库匹配。bio-variant-normalization 技能使用 bcftools norm -m-any 分解多等位基因位点并将等位基因相对于参考基因组进行左对齐。bio-vcf-statistics 技能计算转换/颠换比值——全基因组测序的一项关键质量指标,预期值约为 2.0–2.1——以及用于质量评估的杂合率和深度分布 。
4.7 临床解读
原始变异注释与可操作的临床洞察之间的桥梁由两个技能构成:bio-variant-calling-clinical-interpretation 和 bio-variant-calling-variant-prioritization。这些技能应用 ACMG/AMP 指南对变异致病性进行分类,并通过整合来自人群数据库、计算预测、功能数据和临床文献的证据来对候选变异进行排序。临床解读层由 11 个专用的临床数据库技能提供支持,这些技能提供了底层的证据来源。
bio-clinical-databases-clinvar-lookupbio-clinical-databases-dbsnp-queriesbio-clinical-databases-gnomad-frequenciesbio-clinical-databases-myvariant-queriesbio-clinical-databases-pharmacogenomicsbio-clinical-databases-polygenic-riskbio-clinical-databases-somatic-signaturesbio-clinical-databases-tumor-mutational-burdenbio-clinical-databases-variant-prioritizationbio-clinical-databases-hla-typingbio-clinical-databases-cfdna-preprocessing除了 SNP 和小型插入缺失外,变异检测领域还扩展到了结构变异和体细胞突变。bio-variant-calling-structural-variant-calling 技能处理来自长读长或双端测序数据的大规模基因组重排,涵盖检测、分类和致病性评估。体细胞分析由五个专用技能集群提供服务:bio-copy-number-cnvkit-analysis、bio-copy-number-gatk-cnv、bio-tumor-fraction-estimation、bio-ctdna-mutation-detection 和 bio-longitudinal-monitoring——它们共同实现了液体活检和肿瘤演化追踪工作流。
要使用这些技能,请将它们安装到你的 OpenClaw 或 NanoClaw 工作区中。变异检测和注释技能分布在 skills/ 目录下,带有 bio-variant-* 和 bio-vcf-* 前缀。VARIANT_CALLING_SKILLS=( "bio-variant-calling" "bio-gatk-variant-calling" "bio-variant-calling-deepvariant" "bio-variant-calling-joint-calling" "bio-variant-calling-structural-variant-calling" "bio-variant-calling-filtering-best-practices")# 注释层ANNOTATION_SKILLS=( "bio-variant-annotation" "bio-variant-normalization" "bio-variant-calling-clinical-interpretation" "bio-clinical-databases-clinvar-lookup" "bio-clinical-databases-gnomad-frequencies" "bio-clinical-databases-myvariant-queries")# VCF 实用工具VCF_SKILLS=( "bio-vcf-basics" "bio-vcf-manipulation" "bio-vcf-statistics")ALL_SKILLS=("${VARIANT_CALLING_SKILLS[@]}" "${ANNOTATION_SKILLS[@]}" "${VCF_SKILLS[@]}")for skill in "${ALL_SKILLS[@]}"; do cp -r OpenClaw-Medical-Skills/skills/$skill ~/.openclaw/skills/done
📌 往期回顾
第1期:开源医疗AI的“武器库”OpenClaw Medical Skills:869个技能,拿走不谢
第二期:OpenClaw Medical Skills 生信技能详解|质控与读长处理
第三期:OpenClaw Medical Skills 生信技能详解|变异检测与注释
📢 下期预告:《OpenClaw Medical Skills 生信技能详解|差异表达与转录组学》,敬请期待
附1 variant-calling技能软件汇总:
| | |
| | 从BAM文件生成每碱基位置的基因型似然值(pileup),是变异calling的第一步;支持质控过滤、区域限定、注释标签(DP/AD等)输出 |
| | 从mpileup生成的基因型似然值中调用SNP和Indel;支持多等位基因caller(-m,推荐)和旧版共识caller(-c);可指定倍性 |
| | 合并按染色体分块并行calling后的多个VCF文件 |
| | 通过局部从头单倍型组装(de novo assembly)检测胚系SNP和Indel,是GATK最佳实践的核心calling工具;支持单样本VCF模式和多样本GVCF模式 |
| | 碱基质量评分重校准(BQSR)第一步:在已知变异位点建立碱基质量误差模型,生成校准表 |
| | BQSR第二步:将BaseRecalibrator生成的校准表应用于BAM,输出校准后的BAM文件 |
| | 大型队列(>100样本)gVCF合并:将多个per-sample gVCF导入GenomicsDB数据库,支持按染色体并行、增量更新新样本 |
| | 小型队列(<100样本)gVCF合并:将多个per-sample gVCF合并为一个cohort gVCF |
| | 联合基因分型:从CombineGVCFs输出或GenomicsDB数据库中对所有样本进行联合基因型推断,生成最终cohort VCF |
| | 变异质量评分重校准(VQSR)模型训练:利用HapMap/Omni/1000G/dbSNP/Mills等已知变异集,基于高斯混合模型对SNP或Indel质量注释建模 |
| | 应用VQSR模型:根据指定truth sensitivity阈值(SNP 99.5%,Indel 99.0%)对变异进行过滤,在FILTER列标注 |
| | 从VCF中分离特定类型的变异(SNP或Indel),用于后续分型别的硬过滤 |
| | 硬过滤:按QD/FS/MQ/SOR/MQRankSum/ReadPosRankSum等注释阈值为变异添加FILTER标签(适用于小样本/外显子组/无法使用VQSR的情况) |
| | 合并过滤后的SNP和Indel VCF文件为一个完整VCF |
| | 基于卷积神经网络(CNN)对变异进行评分,是VQSR的深度学习替代方案 |
| GATK FilterVariantTranches | 根据CNNScoreVariants评分,按sensitivity阈值过滤变异 |
| bio-variant-calling-deepvariant | DeepVariant (run_deepvariant) | Google开发的深度学习变异calling工具:将reads pileup转为图像张量,经CNN分类输出基因型VCF;支持Illumina WGS/WES、PacBio HiFi和ONT数据,精度业界最高 |
| bio-variant-calling-deepvariant | | DeepVariant gVCF的多样本联合基因分型工具;替代GATK GenotypeGVCFs,专为DeepVariant输出设计,支持WGS/WES配置 |
| bio-variant-calling-deepvariant | | 对照GIAB(Genome in a Bottle)truth set评估变异calling准确性,计算SNP和Indel的精确率(Precision)、召回率(Recall)、F1 |
| bio-variant-calling-deepvariant | | DeepVariant输出变异的统计分析与质量过滤(Ti/Tv比值评估、GQ/QUAL过滤) |
| bio-variant-calling-joint-calling | GATK HaplotypeCaller (GVCF模式) | 以-ERC GVCF模式生成per-sample gVCF文件,记录所有位点(含参考置信区间),用于后续多样本联合基因分型 |
| bio-variant-calling-joint-calling | | 小型队列(<100样本)gVCF合并,将多个per-sample gVCF合并为单个cohort gVCF |
| bio-variant-calling-joint-calling | | 大型队列(>100样本)gVCF导入GenomicsDB:支持按染色体并行分散处理、增量追加新样本,适合超大规模队列 |
| bio-variant-calling-joint-calling | | 联合基因分型:对所有样本进行统一的基因型推断,可选加入等位基因特异性注释(AS_StandardAnnotation) |
| bio-variant-calling-joint-calling | GATK VariantRecalibrator + ApplyVQSR | 队列联合calling后的VQSR过滤(适用于>30样本):分别对SNP和Indel进行机器学习质量评分重校准 |
| bio-variant-calling-joint-calling | | |
| bio-variant-calling-structural-variant-calling | | 短读长结构变异(SV)calling工具(推荐):基于split-read和paired-end信号检测缺失/插入/倒位/重复/易位;支持胚系、肿瘤-正常对(体细胞)、外显子组和RNA-seq模式 |
| bio-variant-calling-structural-variant-calling | | 短读长SV calling工具:支持多样本联合calling及肿瘤-正常对体细胞SV过滤;可按SV类型(DEL/DUP/INV/INS/BND)单独calling |
| bio-variant-calling-structural-variant-calling | | 基于split-read和discordant pair证据的SV calling工具;lumpyexpress为简化版封装 |
| bio-variant-calling-structural-variant-calling | | LUMPY的简化封装工具:自动化split-read提取和discordant pair处理,简化LUMPY运行流程 |
| bio-variant-calling-structural-variant-calling | | 多SV calling工具结果合并:按断点距离和SV类型一致性要求,合并Manta/Delly/LUMPY等多个callset,提高SV置信度 |
| bio-variant-calling-structural-variant-calling | | 结构变异注释工具:整合基因组注释(基因、外显子)、DGV、gnomAD-SV、ClinVar等数据库,对SV进行功能和致病性注释 |
| bio-variant-calling-structural-variant-calling | | SV结果过滤:按质量值、SVLEN、SVTYPE和FILTER状态筛选结构变异 |
| bio-variant-calling-structural-variant-calling | | LUMPY前置步骤:提取discordant pairs(插入片段异常的read对)和split reads(跨越SV断点的reads) |
| bio-variant-calling-filtering-best-practices | | SNP和Indel硬过滤:按QD/FS/MQ/SOR/MQRankSum/ReadPosRankSum等注释阈值为变异添加FILTER标签(适用于小样本/外显子组/VQSR不可用场景) |
| bio-variant-calling-filtering-best-practices | | VQSR模型训练(适用于>30个外显子组或WGS大队列):利用HapMap/Omni/1000G/dbSNP/Mills等已知变异集,按QD/MQ/FS/SOR/RankSum等注释构建高斯混合模型 |
| bio-variant-calling-filtering-best-practices | | 应用VQSR模型:根据truth sensitivity阈值(SNP 99.5%,Indel 99.0%)过滤变异,在FILTER列添加标签 |
| bio-variant-calling-filtering-best-practices | | 体细胞变异专用过滤:结合污染率估计表和肿瘤分割信息,过滤Mutect2输出的原始变异 |
| bio-variant-calling-filtering-best-practices | | 基于表达式的变异软过滤/硬过滤:支持QUAL/INFO/FORMAT字段的复杂逻辑表达式,可添加命名FILTER标签(软过滤)或直接删除变异(硬过滤) |
| bio-variant-calling-filtering-best-practices | | 按变异类型、基因组区域、样本子集筛选变异;提取PASS变异;双等位基因过滤 |
| bio-variant-calling-filtering-best-practices | | Python编程式变异过滤:逐变异遍历VCF,按QUAL/INFO/FORMAT字段自定义多条件过滤逻辑,输出过滤后VCF |
| bio-variant-calling-filtering-best-practices | | 过滤效果验证:统计过滤前后变异数量、Ti/Tv比值变化(WGS期望~2.1,外显子组2.8-3.3),评估过滤质量 |
附2 variant-annotation技能软件汇总:
| | |
| | 向VCF添加或移除注释字段:从dbSNP/ClinVar等数据库VCF、BED或TAB文件中按位置匹配,复制ID/INFO等字段;也可用--set-id生成自定义变异ID |
| | 基于GFF3基因模型对变异进行简单功能后果预测,输出synonymous/missense/stop_gained/frameshift/splice等分类,速度快,适合初步注释 |
| VEP (Ensembl Variant Effect Predictor) | 全面的变异功能注释工具(金标准):预测编码后果、影响级别(HIGH/MODERATE/LOW/MODIFIER)、HGVS命名、SIFT/PolyPhen致病性预测、gnomAD频率、ClinVar,支持CADD/dbNSFP/SpliceAI等丰富插件 |
| | 快速批量变异功能注释工具:将变异按HIGH/MODERATE/LOW/MODIFIER四个影响级别分类,生成统计HTML报告,ANN字段包含基因名/HGVS等信息 |
| | SnpEff配套过滤与数据库注释工具:支持dbSNP/ClinVar/dbNSFP注释,以及基于表达式的VCF过滤(按QUAL/DP/CLNSIG等字段) |
| | 灵活的多数据库变异注释工具:通过-protocol参数组合refGene(基因注释)、gnomAD(频率)、ClinVar(临床意义)、dbNSFP(功能预测)等多种注释协议 |
| | Python解析VEP CSQ字段或SnpEff ANN字段的管道符分隔注释,按影响级别过滤变异并提取基因名、后果类型、HGVS等信息 |
| bio-variant-normalization | | 变异标准化一体化工具:Indel左对齐(-f)、多等位基因位点拆分/合并(-m)、REF等位基因校正(-c)、MNP原子化分解(--atomize)、精确重复变异去除(-d);标准化是数据库查询和多caller比较的必要前置步骤 |
| bio-variant-normalization | | 标准化后的多caller VCF比较:按位置取交集/并集/差集,用于评估不同caller的一致性 |
| bio-variant-normalization | | Python预检变异标准化需求:统计VCF中多等位基因位点和MNP数量,评估是否需要在bcftools norm之前进行标准化处理 |
| bio-variant-calling-clinical-interpretation | | 按ACMG/AMP 2015指南自动评估28条致病性分类标准(PVS1/PS1-4/PM1-6/PP1-5/BA1/BS1-4/BP1-7),输出Pathogenic/Likely pathogenic/VUS/Likely benign/Benign五级分类 |
| bio-variant-calling-clinical-interpretation | bcftools annotate + bcftools view | ClinVar临床意义注释及致病性过滤:导入CLNSIG/CLNDN/CLNREVSTAT字段,按致病性分类和审查星级(0-4星)筛选高置信度致病/可能致病变异 |
| bio-variant-calling-clinical-interpretation | | Python多维度临床优先级分类:整合ClinVar致病性、gnomAD频率、CADD和REVEL评分,按PATHOGENIC/LIKELY_PATHOGENIC/VUS/BENIGN分级输出优先变异列表 |
| bio-variant-calling-clinical-interpretation | | 生成临床报告TSV文件:提取CHROM/POS/REF/ALT/基因名/功能后果/CLNSIG/CLNDN/gnomAD_AF/CADD_PHRED等字段输出为表格 |
| bio-clinical-databases-clinvar-lookup | requests (Python) + NCBI E-utilities | 通过NCBI E-utilities REST API在线查询ClinVar:支持按变异ID(esummary)、基因名(esearch[gene])、HGVS表示法查询致病性分类和疾病关联 |
| bio-clinical-databases-clinvar-lookup | cyvcf2 (Python) — 本地ClinVar VCF | 本地ClinVar VCF离线查询:按染色体坐标+REF/ALT精确匹配,返回CLNSIG/CLNREVSTAT/CLNDN/CLNVC字段,速度快,适合批量样本处理 |
| bio-clinical-databases-clinvar-lookup | bcftools annotate (批量ClinVar注释) | 批量将ClinVar临床字段注入整个VCF文件,高效处理大规模变异集,支持按CLNSIG/CLNREVSTAT过滤高置信度致病变异 |
| bio-clinical-databases-gnomad-frequencies | requests (Python) — gnomAD GraphQL API | 通过gnomAD REST API(GraphQL)查询单个变异的外显子组/基因组等位基因频率(AF/AC/AN/纯合子数),支持gnomad_r4/r3/r2_1数据集版本 |
| bio-clinical-databases-gnomad-frequencies | myvariant (Python) — gnomAD频率 | 通过myvariant.info聚合接口查询gnomAD外显子组/基因组频率及人群分层频率(AFR/AMR/ASJ/EAS/FIN/NFE/SAS),比直接GraphQL更简洁 |
| bio-clinical-databases-gnomad-frequencies | Hail (Python) — 本地gnomAD大规模查询 | 使用Hail框架加载gnomAD Hail Table(Google Cloud Storage),对大规模变异集进行高效频率过滤,适用于全基因组级别分析 |
| bio-clinical-databases-myvariant-queries | | myvariant.info聚合API:一次请求从ClinVar/gnomAD/dbSNP/COSMIC/CADD/dbNSFP/SnpEff等多数据库获取聚合注释;支持单变异查询、批量查询(≤1000变异/次)、基因/区域搜索;结果直接转DataFrame |
附3 vcf实用工具技能软件汇总:
| | |
| | VCF/BCF文件查看、格式转换与子集提取:支持查看全文/仅header/去header,按区域/样本子集显示,输出VCF/VCF.gz/BCF/BCF.gz多种格式 |
| | 按自定义格式提取VCF字段为制表符分隔文本:支持CHROM/POS/ID/REF/ALT/QUAL/FILTER/INFO字段及样本级FORMAT字段(GT/DP/GQ等) |
| | VCF文件压缩与索引:bgzip产生可随机访问的bgzf压缩格式(非普通gzip),bcftools index生成CSI或tabix(TBI)索引,区域查询必需 |
| | Python编程式VCF读写库:高性能迭代变异记录,以属性方式访问CHROM/POS/REF/ALT/QUAL/FILTER/INFO/FORMAT字段及基因型矩阵,支持区域查询和过滤后写出 |
| | 多样本VCF合并:将多个含不同样本列的VCF合并为单个多样本VCF(相同位置对齐),缺失基因型默认填./. 或可选填0/0;支持从文件列表批量合并 |
| | 区域VCF合并:将相同样本集按不同基因组区域(如按染色体分块)的多个VCF拼接为完整VCF;支持允许区域重叠(-a)和去重(-d) |
| | VCF文件按染色体和位置排序;支持大文件临时目录和内存限制设置 |
| | 多VCF取交集/差集/比较:输出各文件私有变异和共有变异子集;支持按出现文件数量(-n)或布尔掩码(-n~)精确控制 |
| | 重命名VCF中的样本列名:通过映射文件批量修改样本名称,用于样本名标准化或错误修正 |
| | 从多样本VCF提取样本子集或按区域提取,也可按染色体循环拆分为多个独立VCF文件 |
| | VCF全面质量统计工具:输出SN(变异数量汇总)、TSTV(转换/颠换比值)、AF(等位基因频率分布)、QUAL(质量分布)、IDD(Indel长度分布)、DP(测序深度分布)等多节统计;支持单样本统计、多VCF比较和区域限定 |
| | 将bcftools stats输出可视化为PDF/PNG图表,包括变异类型分布、质量分布、Indel长度分布等,用于质控报告生成 |
| | 样本基因型一致性验证:计算样本间两两差异率(discordance rate),用于检测样本交叉污染、样本置换错误或与已知参考面板比对验证身份 |
| bcftools query + awk (快速统计) | bcftools query提取字段后结合awk进行自定义统计:变异计数、PASS变异数、质量均值、深度均值、等位基因频率谱、杂合子计数等 |
| | Python编程式变异统计:自定义SNP/Indel计数、质量分布(numpy)、基因型分布(HOM_REF/HET/HOM_ALT/缺失)、Ti/Tv比值计算 |
 夜雨聆风
夜雨聆风