 夜雨聆风
夜雨聆风TCGA数据下载和处理
https://portal.gdc.cancer.gov/

基本介绍
TCGA(癌症基因组图谱)是美国国家癌症研究所(NCI)与美国国家人类基因组研究所(NHGRI)联合发起的大规模癌症研究项目。截至 GDC Data Release 45.0(2025 年 12 月 4 日),基因组数据公共库(Genomic Data Commons, GDC)已累计收录50,270 例病例(cases),覆盖69 个原发癌部位,包含 TCGA 等多个核心癌症基因组研究项目的临床信息、基因组变异、转录组(mRNA/miRNA)、甲基化等多维度组学数据,是全球癌症基础与转化研究的核心数据资源。其中 TCGA 的全部数据已完整整合至 GDC 平台,可通过 GDC Data Portal 网页端进行检索、筛选与下载。为满足不同数据的获取需求,本文将从三个方面进行介绍:TCGA 网页端直接下载、UCSC 网页端快速获取预处理数据,以及基于 TCGAbiolinks 包的程序化批量下载,为后续分析提供标准化、可重复的数据获取流程。
01
TCGA网页端下载数据
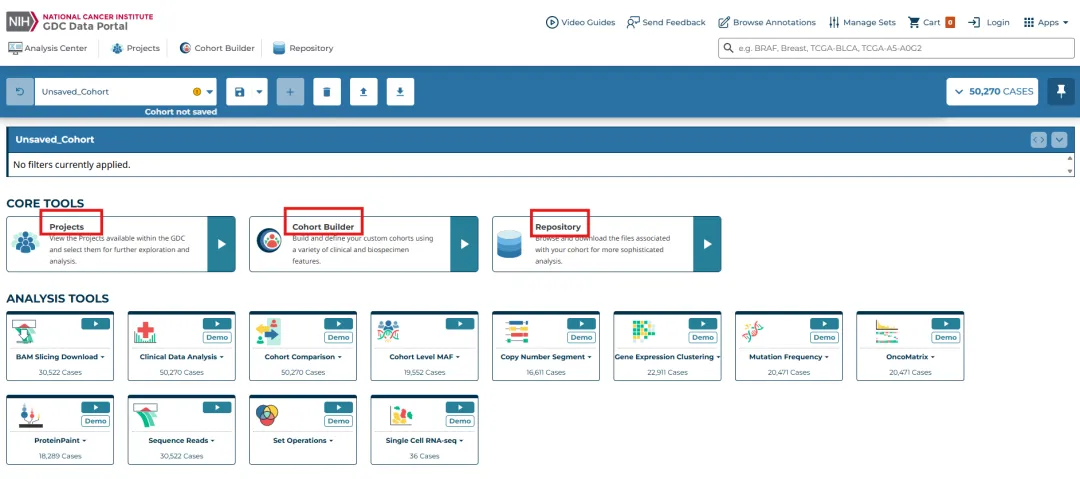
进行主页面,包括三个部分,Projects(浏览定位目标癌症研究项目)→Cohort Builder(在项目中筛选符合研究需求的样本队列)→Repository(针对筛选后的样本,选择并下载具体数据文件)
1
Projects
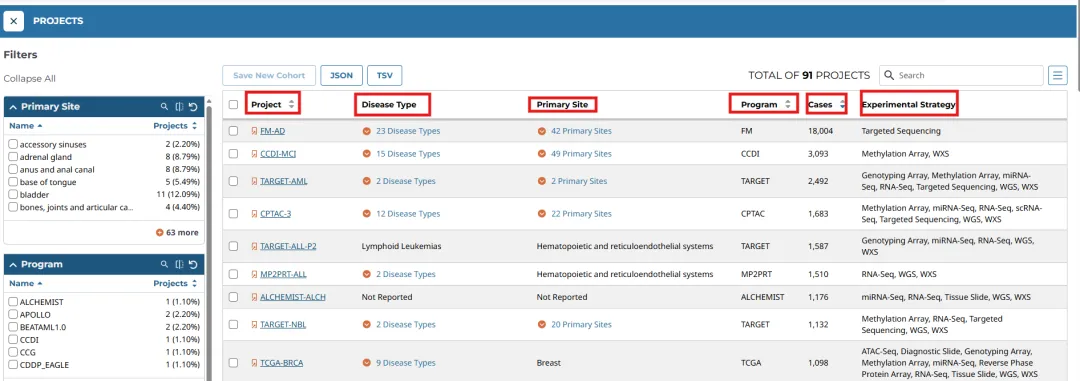
Projects中主要包括五个部分:
Primary Site(原发部位)
Program(项目)
Disease Type(疾病类型)
Data Category(数据类别)
Experimental Strategy(测序方法)
项目中不仅仅包括TCGA数据,还包括TARGET(青少年难治性癌症基因组计划)、CGCI(特殊癌症基因组补充计划)等其他相关癌症数据。
2
Cohort Builder
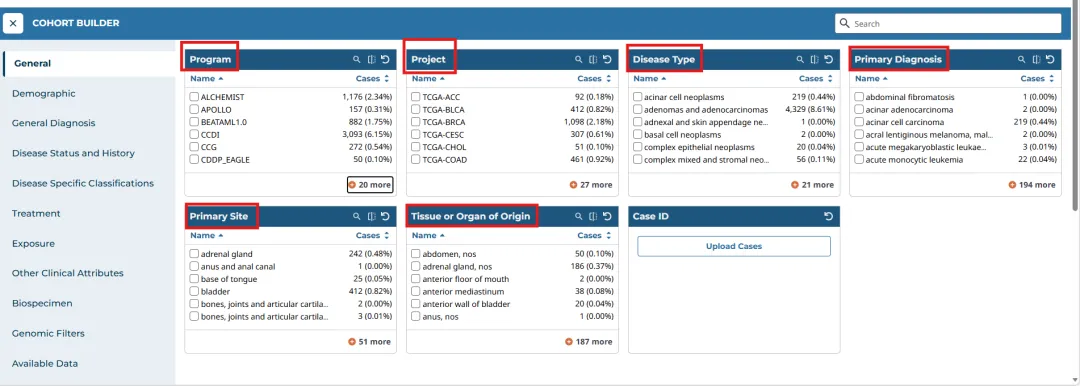
Cohort Builder中主要包括六个部分:
Program(项目计划)
Project(具体项目)
Disease Type(疾病类型)
Primary Diagnosis(主要诊断)
Primary Site(原发部位)
Tissue or Organ of Origin(组织、器官)
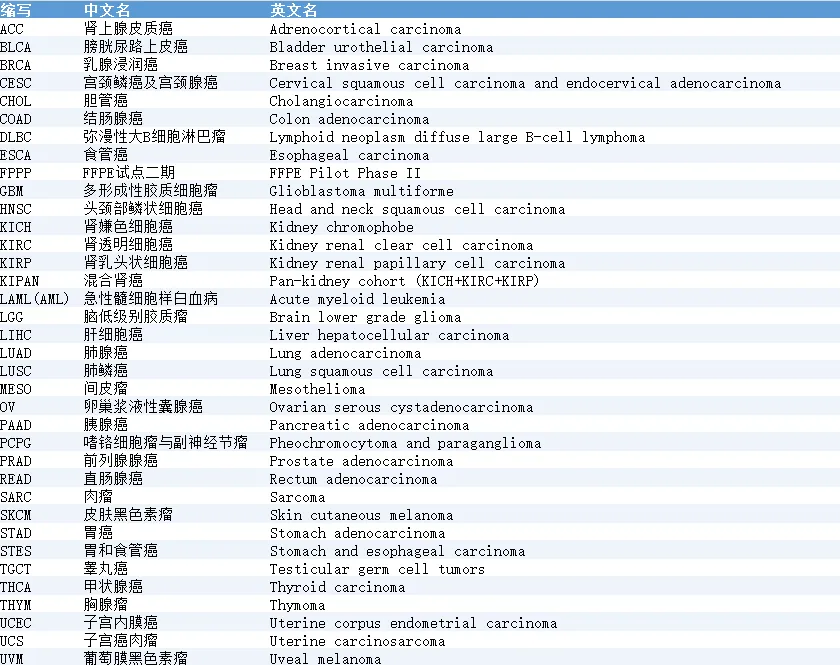
下面是TCGA中肿瘤类型对照表:
本次我们以TCGA-LUAD肺腺癌作为示例,在当前页面选择:
Program=TCGA
Project=TCGA-LUAD
然后切换到Repository页面继续检索⬇️⬇️
3
Repository
切换到Repository页面之后可以发现,我们已经选择了TCGA-LUAD,接下来进行细化检索,其中常见的选择包括:
Experimental Strategy(实验策略): RNA‑Seq
Data Category(数据类型): Transcriptome Profiling
Data Type(具体数据类型):
Gene Expression Quantification
Access(数据权限): Open
Tissue Type:normal/tumor
Tumor Descriptor(肿瘤描述):
Primary Tumor
在选择之后,可以看到从 TCGA 数据库获取肺腺癌(LUAD)原发肿瘤样本的公开 RNA‑Seq 基因表达定量数据,共包含601 个表达文件,其中肿瘤组织 542 例、正常组织 59 例,数据总量 2.55 GB。
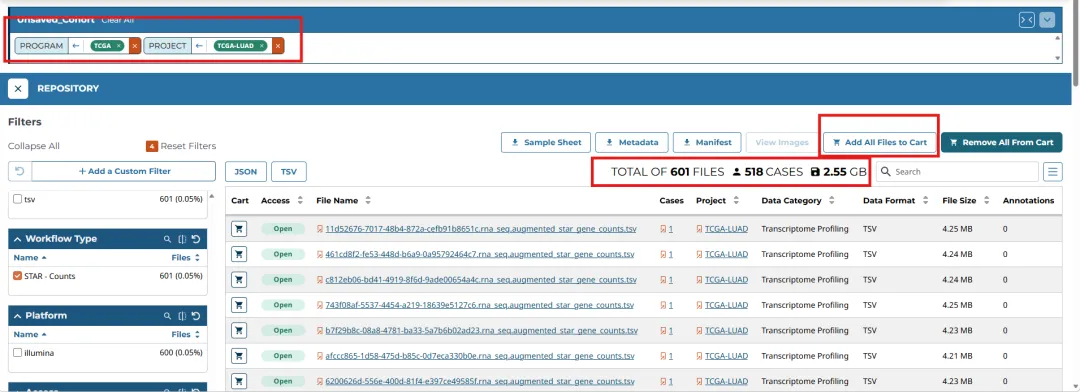
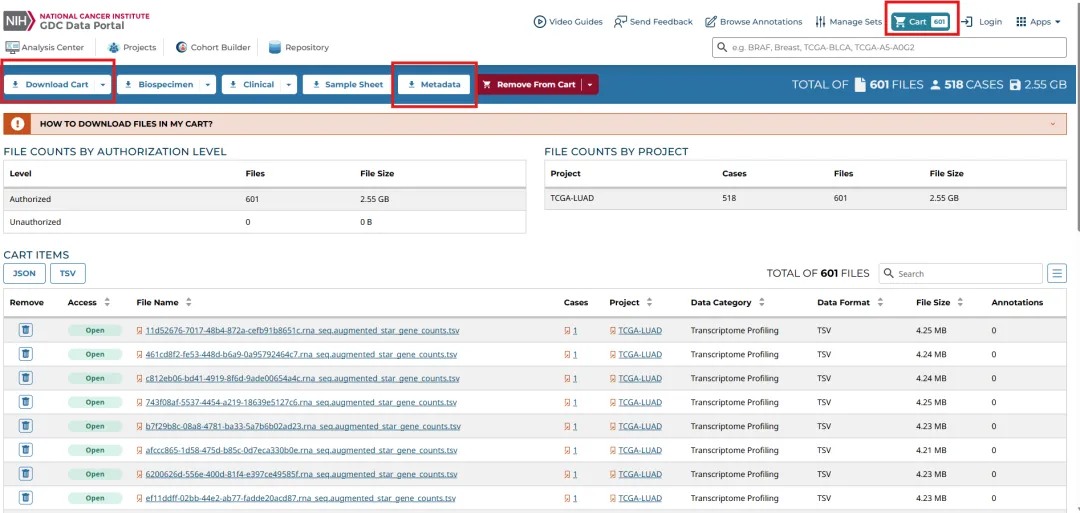
在选择好数据之后点击"Add All Files to Cart"。将所有文件添加至Cart,然后切换到Cart页面进行数据下载。下载好后就可进行整理使用啦!
02
UCSC网页端下载数据
通过浏览器检索UCSC xena,进入官网(http://xena.ucsc.edu/)点击Launch Xena,随后点击左上角DATA SETS或直接点击链接进入UCSC数据页面https://xenabrowser.net/datapages/
1
查找并下载数据
选择要下载的TCGA数据,以TCGA-LAML数据为例,点击GDC TCGA Acute Myeloid Leukemia (LAML) 进入相关页面
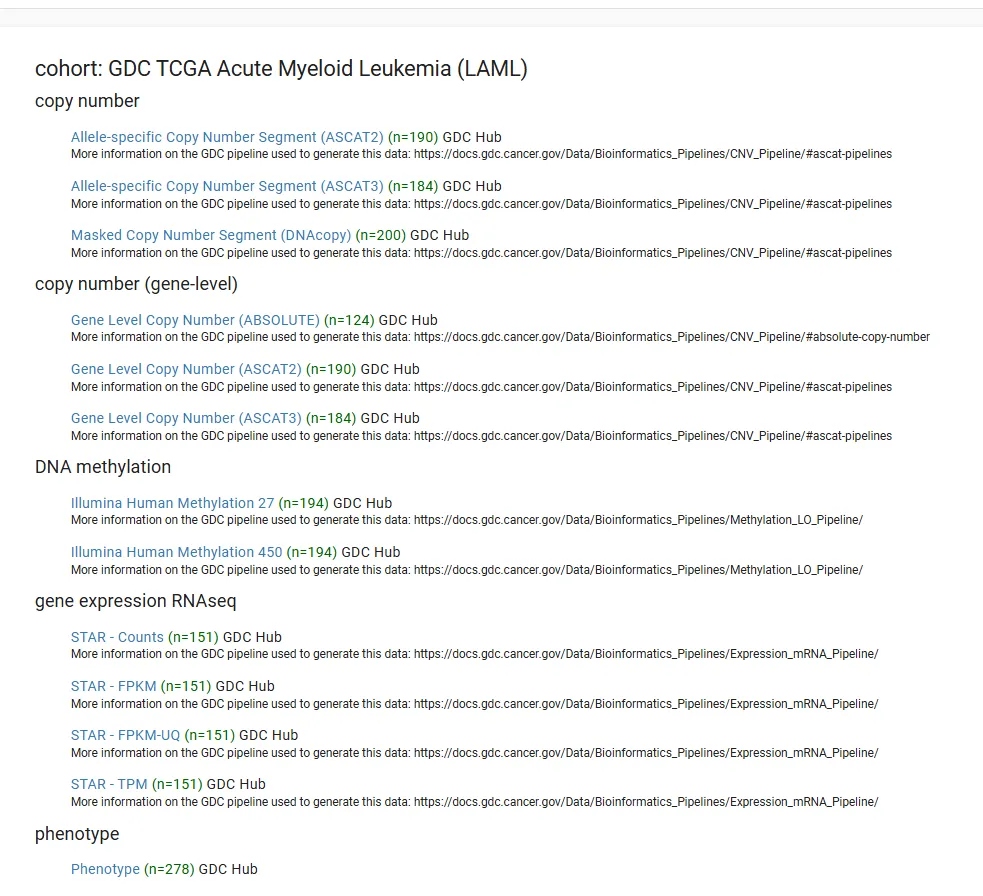
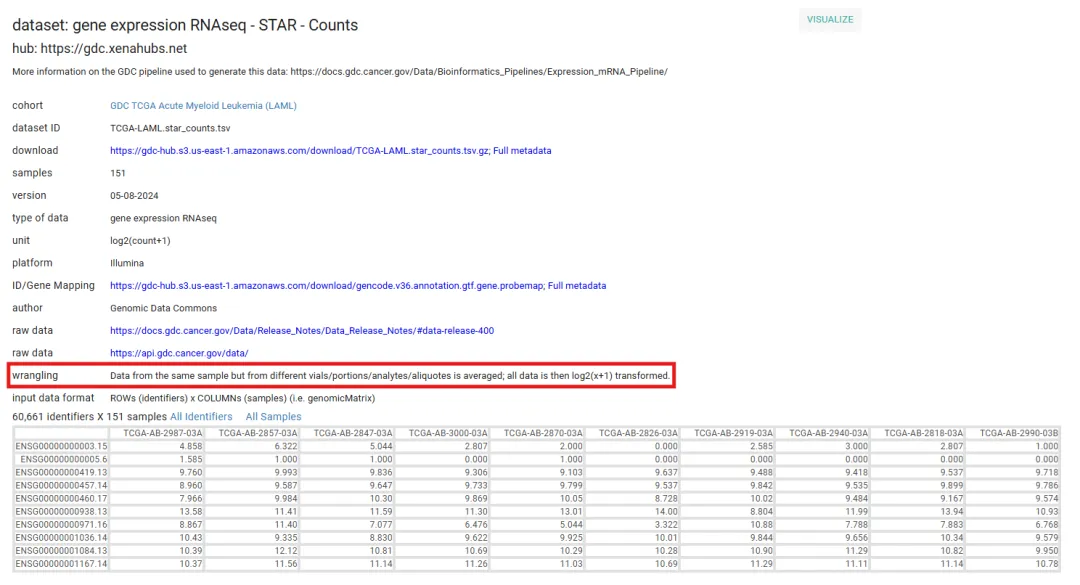
可以看到多种数据类型,其中gene expression RNAseq数据为转录组数据(包含原始表达数据STAR - Counts和处理后FPKM、TPM等)。phenotype包含Phenotype(样本各种数据包括年龄、性别、病史、阶段等)和survival data(生存数据)。
!!注:UCSC xena中的counts、fpkm、tpm数据是log2(counts+1)、log2(fpkm+1)、log2(tpm+1)的结果。
2
处理数据
以上述查找的TCGA-LAML数据为例,下载其转录组测序数据以及其临床数据信息解压至本地进行处理
library(data.table)library(dplyr)library(tidyr)library(tibble)library(limma)library(DESeq2)# TPM数据的处理 -----------------------------------------------------------logtpm<- fread("TCGA-LAML.star_tpm.tsv",data.table = F)logtpm$Ensembl_ID<- substr(logtpm$Ensembl_ID,1,15)logtpm <- as.data.frame(avereps(logtpm[,-1], ID = logtpm$Ensembl_ID))##去除在所有样本中不表达的基因logtpm <- logtpm[apply(logtpm, 1,function(x)sum(x!=0)>0),]# FPKM数据的处理 -----------------------------------------------------------logfpkm<- fread("TCGA-LAML.star_fpkm.tsv",data.table = F)logfpkm$Ensembl_ID<- substr(logfpkm$Ensembl_ID,1,15)logfpkm <- as.data.frame(avereps(logfpkm[,-1], ID = logfpkm$Ensembl_ID))##去除在所有样本中不表达的基因logfpkm <- logfpkm[apply(logfpkm, 1,function(x)sum(x!=0)>0),]# 临床数据的处理-------------------------------------------------------------------------#读取生存数据sur <- fread("TCGA-LAML.survival.tsv",data.table = F)sur$OS.time<- as.numeric(sur$OS.time)sur$OS<- as.numeric(sur$OS)rownames(sur) <- sur$sample#读取临床数据cli <- fread("TCGA-LAML.clinical.tsv",data.table = F)cli <- cli[, c("sample","primary_site","disease_type","gender.demographic","race.demographic","age_at_index.demographic","age_at_diagnosis.diagnoses","vital_status.demographic","days_to_death.demographic","days_to_last_follow_up.diagnoses","sample_type.samples","specimen_type.samples")]#将生存数据与临床数据合并TCGA_aml <- merge(sur,cli,by=1)table(TCGA_aml$OS)TCGA_aml$OS.time<- as.numeric(TCGA_aml$OS.time)/365TCGA_aml$Age<- as.numeric(TCGA_aml$age_at_diagnosis.diagnoses)table(TCGA_aml$gender.demographic)TCGA_aml$Gender<- ifelse(TCGA_aml$gender.demographic=='female','Female','Male')TCGA_aml2 <- TCGA_aml%>% dplyr::select(sample,OS,OS.time,Age,Gender,everything())#临床数据与表达数据合并x <- intersect(TCGA_aml2$sample,colnames(logtpm))TCGA_aml2 <- TCGA_aml2[TCGA_aml2$sample%in%x,]logfpkm <- logfpkm[,x]logtpm <- logtpm[,x]#保存数据save(logfpkm,logtpm,TCGA_aml2,file ='TCGA_LAML.rda')
03
使用TCGAbiolinks包进行数据下载
TCGAbiolinks是 R 语言中专门用于下载、整理、分析 TCGA数据的核心生物信息学包,整合 GDC官方 API,解决了 TCGA 原始数据下载繁琐、格式杂乱、预处理复杂的痛点。
if(!require("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("TCGAbiolinks")
可以查看R包中共有多少种TCGA数据:
library(TCGAbiolinks)projects <- TCGAbiolinks::getGDCprojects()$project_idprojects <- projects[grepl('^TCGA', projects, perl=TRUE)]projects[1] "TCGA-LGG" "TCGA-BRCA""TCGA-LAML""TCGA-UCS" "TCGA-GBM"[6] "TCGA-THYM""TCGA-TGCT""TCGA-PCPG""TCGA-CHOL""TCGA-DLBC"[11] "TCGA-CESC""TCGA-ESCA""TCGA-ACC" "TCGA-KICH""TCGA-HNSC"[16] "TCGA-READ""TCGA-COAD""TCGA-BLCA""TCGA-LIHC""TCGA-MESO"[21] "TCGA-KIRP""TCGA-SKCM""TCGA-SARC""TCGA-LUAD""TCGA-PRAD"[26] "TCGA-LUSC""TCGA-PAAD""TCGA-UVM" "TCGA-KIRC""TCGA-THCA"[31] "TCGA-OV" "TCGA-UCEC""TCGA-STAD"
数据查询GDCquery():构建查询,指定癌症、数据类型、样本类型、平台等,不直接下载,仅返回检索结果。
project:TCGA项目如TCGA-LAML,TCGA-BRCA等
data.category:数据大类(Transcriptome Profiling,DNA Methylation,Simple Nucleotide Variation)
data.type:数据子类(Gene Expression Quantification、Methylation Beta Value等)
workflow.type:分析流程(HTSeq - FPKM、HTSeq - Counts、STAR - Counts)
sample.type:样本类型(Primary Tumor原发肿瘤、Solid Tissue Normal癌旁正常)
query<- GDCquery(project = "TCGA-READ",data.category = "Transcriptome Profiling",data.type = "Gene Expression Quantification",workflow.type = "STAR - Counts",)getResults(query)
数据下载GDCdownload():基于GDCquery的结果,自动下载所有匹配文件,支持断点续传、本地缓存。
GDCdownload(query)Downloading data for project TCGA-READGDCdownload will download 177 files. A total of 747.57671 MBDownloading as: Wed_Apr__8_16_04_30_2026.tar.gzDownload failed. We will retry with smaller chunksDownloading chunk 1 of 8 (24 files, size = 101.244325 MB) as Wed_Apr__8_16_04_30_2026_0.tar.gz
数据整理GDCprepare():将下载的原始文件(BAM、TSV 等)转换为SummarizedExperiment 对象(包含表达矩阵、样本信息、基因注释)。
基因 ID 转换(Ensembl ID ↔ Gene Symbol)
样本去重、过滤低质量样本
整合临床表型数据(年龄、性别、生存时间、分期等)
生成标准化表达矩阵(行 = 基因,列 = 样本)
# 整理数据data <- GDCprepare(query)|===============================================|100% Completed after 6 sStarting to add information to samples=> Add clinical information to samples=> Adding TCGA molecular information from marker papers=> Information will have prefix 'paper_'read subtype information from:doi:10.1038/nature11252Available assays in SummarizedExperiment :=> unstranded=> stranded_first=> stranded_second=> tpm_unstrand=> fpkm_unstrand=> fpkm_uq_unstrand# 提取基因表达矩阵(counts)counts <- assay(data, "unstranded")tpm <- assay(data, "tpm_unstrand")fpkm <- assay(data, "fpkm_unstrand")# 提取样本临床信息clinical_data <- colData(data)# 提取基因注释(ID、symbol、类型)gene_anno <- rowData(data)#样本信息Group <- make_tcga_group(counts)table(Group)#如果处理的数据为AML数据,和其他TCGA数据有一点区别,因此在使用这个函数时会报错,显示临床数据中缺失`disease_response`列data <- GDCprepare(query)|==============================================|100% Completed after 4 sStarting to add information to samples=> Add clinical information to samplesError in `dplyr::select()`:! Can't select columns that don't exist.✖ Column `disease_response` doesn't exist.Run `rlang::last_trace()` to see where the error occurred.
临床数据处理GDCquery_clinic:下载整理 TCGA 临床数据(生存、病理分期、治疗信息等),无需关联组学数据。
clinical <- GDCquery_clinic(project = "TCGA-READ", type = "clinical")dim(clinical)[1] 172157head(clinical[,c("bcr_patient_barcode","vital_status","days_to_death","days_to_last_follow_up")])bcr_patient_barcode vital_status days_to_death days_to_last_follow_up1 TCGA-AF-3912 Not Reported NA NA2 TCGA-AG-4009 Alive NA 4263 TCGA-G5-6572 Dead 1432 14324 TCGA-AG-3580 Alive NA 2445 TCGA-AG-3891 Alive NA 5486 TCGA-AF-2693 Alive NA 1155
差异表达分析TCGAanalyze_DEA:内置差异分析流程,支持limma-voom(适合 FPKM/TPM)、edger(适合 raw counts),自动输出差异基因列表(上下调、log2FC、p 值、FDR)。
# 提取两组表达矩阵mat_tumor <- counts[, Group == "tumor"]mat_normal <- counts[, Group == "normal"]# 差异分析(肿瘤 vs 正常)dea_results <- TCGAanalyze_DEA(+ mat1 = mat_tumor,+ mat2 = mat_normal,+ Cond1type = "Tumor",+ Cond2type = "Normal",+ pipeline = "edgeR",+ fdr.cut = 0.05,+ logFC.cut = 1+ )Batch correction skipped since no factors provided----------------------- DEA -------------------------------o 167 samples in Cond1type Tumoro 10 samples in Cond2type Normalo 60660 features as miRNA or genesThis may take some minutes...----------------------- END DEA -------------------------------# 查看差异基因head(dea_results)logFC logCPM PValue FDRENSG00000183034.137.411759 2.0021501.041996e-2416.320748e-237ENSG00000167916.5 7.301191-1.4654107.336898e-1982.225281e-193ENSG00000123560.146.466769 1.6235007.714723e-1791.559917e-174ENSG00000157005.4 6.253946 1.4572664.384521e-1646.649126e-160ENSG00000136546.166.194023 2.3680157.769426e-1639.425867e-159ENSG00000115263.156.150191 3.1783981.070325e-1591.082099e-155# 提取基因注释(包含 ID 和 基因名)gene_anno <- rowData(data)# 给差异结果添加基因名dea_results$gene_id <- rownames(dea_results)dea_results$gene_symbol <- gene_anno[dea_results$gene_id, "gene_name"]deg <- dea_results[, c("gene_symbol", setdiff(colnames(dea_results), "gene_symbol"))]#绘制火山图TCGAVisualize_volcano(x = deg$logFC,y = deg$FDR,names = deg$gene_symbol,x.cut = 1,y.cut = 0.05,filename = "READ_volcano.pdf")
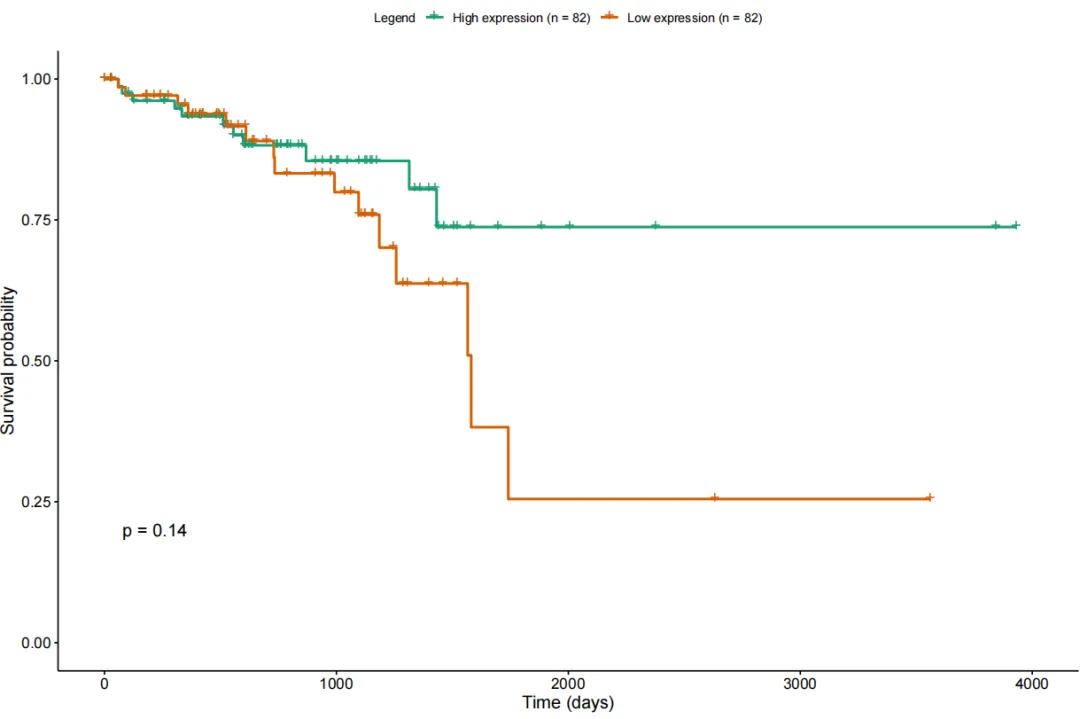
生存分析TCGAanalyze_survival:结合基因表达与临床数据,做Kaplan-Meier 生存分析,自动分组(高表达 / 低表达)、计算 log-rank p 值
library(TCGAbiolinks)library(dplyr)library(tibble)library(survival)library(survminer)expr_surv <- tpmrownames(expr_surv) <- gene_anno$gene_nameexpr_surv <- expr_surv[!duplicated(rownames(expr_surv)), ]tumor_mask <- substr(colnames(expr_surv), 14, 15)== "01"expr_surv <- expr_surv[, tumor_mask]colnames(expr_surv) <- sapply(strsplit(colnames(expr_surv), "-"),function(x) paste0(x[1:3], collapse = "-"))clinical_patient <- GDCquery_clinic(project = "TCGA-READ", type = "clinical")KRAS_expr <- as.data.frame(t(expr_surv["KRAS", , drop = FALSE]))colnames(KRAS_expr) <- "KRAS"KRAS_expr$submitter_id <- rownames(KRAS_expr)df_surv <- clinical_patientdf_surv$KRAS <- KRAS_expr$KRAS[match(df_surv$submitter_id, KRAS_expr$submitter_id)]df_surv <- df_surv[!is.na(df_surv$KRAS), ]df_surv <- df_surv %>%filter(vital_status %in% c("Alive", "Dead")) %>%filter((!is.na(days_to_death) & vital_status == "Dead") |(!is.na(days_to_last_follow_up) & vital_status == "Alive"))df_surv <- df_surv %>%mutate(time = case_when(vital_status == "Dead" ~ days_to_death,vital_status == "Alive" ~ days_to_last_follow_up),event = ifelse(vital_status == "Dead", 1, 0)) %>%filter(!is.na(time))median_expr <- median(df_surv$KRAS, na.rm = TRUE)df_surv$exp_group <- ifelse(df_surv$KRAS >= median_expr,"High expression","Low expression")TCGAanalyze_survival(data = df_surv,clusterCol = "exp_group",timeCol = "time",eventCol = "event",risk.table = FALSE,conf.int = FALSE,color = "Dark2",main = "TCGA-READ KRAS Survival Analysis",xlab = "Time (days)",ylab = "Survival Probability",filename = "READ_KRAS_survival.pdf",pvalue = TRUE)
关于TCGAbiolinks可以点击官方文档了解更详细的内容:https://bioconductor.org/packages/release/bioc/manuals/TCGAbiolinks/man/TCGAbiolinks.pdf
!!!本文所涉及的 TCGA 相关数据统计与获取方法,是基于文章发表前的平台版本与数据发布情况。随着数据库的持续更新与迭代,具体数据信息、平台功能及获取路径请以对应时间的官方最新版本为准。
中科博林
中科博林是一家以“AI虚拟细胞大模型”为核心驱动,可为科研院所、医院及药企客户提供AI药物筛选、多组学测序与分析、类器官建模及试剂研发的高新技术企业,隶属于博林医疗集团,总部位于沈阳博林医疗科技产业园,中心实验室面积1000平方米。自2024年5月正式运营以来,公司持续加大研发投入,截止目前,已获批软件著作权16项,申报专利4项,发现难治型胃癌相关老药新用方案3项,已积累涵盖人肺腺癌、人肺鳞癌、人肺组织、人肝癌、人胃癌、人胃上皮、人乳腺癌、人甲状腺癌、人结肠癌、人软骨、人脑胶质瘤、人前列腺癌、小鼠结肠等十余种样本类型、百余例样本的类器官模型构建经验。核心成果涵盖多组学自动分析流程、AI虚拟细胞大模型、类器官研发体系、难治性疾病老药新用方案等。未来,中科博林将继续以Bioseeker ™AI虚拟细胞大模型为核心,结合多组学及类器官技术,以高效优质的服务态度,在转化医学及新药研发方面更好地为“博林客户”赋能!
