 夜雨聆风
夜雨聆风

从生物信息、蛋白设计到药物发现
一个AI智能体,正在接管整个生物学
BioTender | AI4Bio

生物学研究的碎片化是一个长期未被正视的效率黑洞。结构预测用一套工具,序列设计用另一套,转录组分析又是另一套,数据在工具之间搬来搬去,大量时间消耗在环境配置和格式转换上,而不是真正的科学问题。Biomni Lab 想做的事情很简单——把这些全部装进一个工作台,让 AI 负责工具调度,科学家只需要描述目标。这个想法不新鲜,但他们是第一个把它做成能跑的东西的团队。

CRISPR 奠基人张锋背书!生物人自己的科研 AI 来了——斯坦福团队成立 Phylo!
Biomni Lab 是 Stanford spin-out 公司 Phylo 于 2026 年 2 月正式推出的商业产品,前身是 2025 年 6 月开源的 Biomni 项目,目前已被超过 7000 个实验室和药企采用。平台融资 1350 万美元,a16z 和 Menlo Ventures/Anthropic Anthology Fund 联合领投,科学顾问阵容包括 Carolyn Bertozzi(诺贝尔奖)、张锋(CRISPR)和 Fabian Theis(计算生物学)。
平台链接👉https://biomni.phylo.bio/
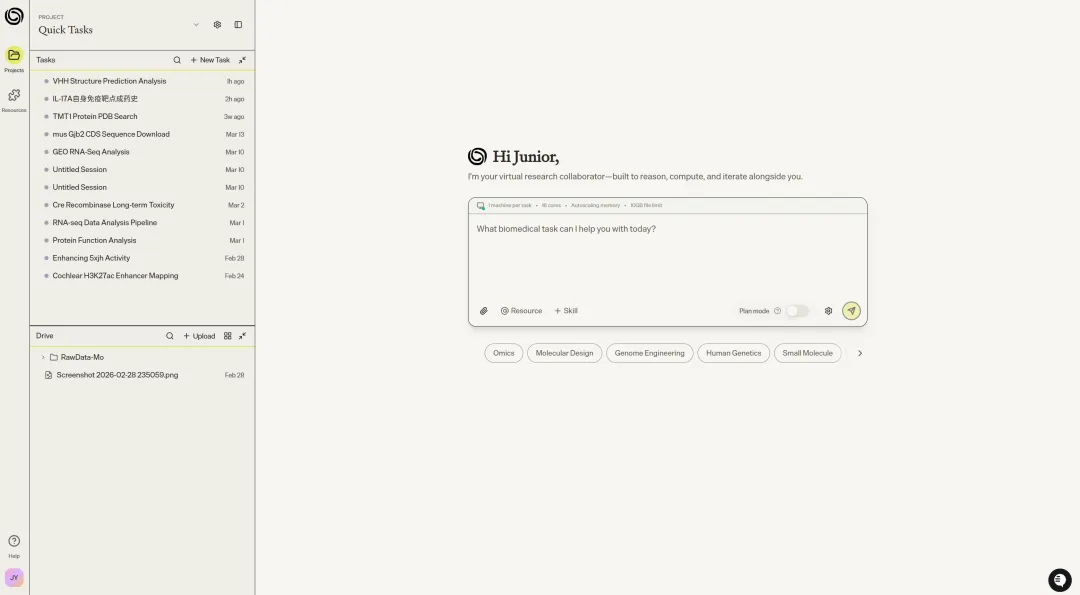
核心定位是"集成生物学环境"(Integrated Biology Environment,IBE)——把 AI Agent、数百种生物信息学工具、59 个生物数据库、105 个生物软件工具整合进一个统一工作台,让科学家用自然语言驱动复杂分析流程。
Biomni Lab往期推荐👇
独家访谈|对话Biomni Lab——重新定义生物学家与AI智能体的交互方式!
AI打通计算与实验最后一公里!PyLabRobot×Biomni重磅!干湿实验不再割裂!!
科研智能体Biomni进化6步:设计蛋白、查数据、跑scRNA、读文献、做假设、超越专家!
Biomni-R0 来了!斯坦福放大招,生物科研专属 AI 直接碾压 GPT-5!
Biomni 0.0.5 重磅更新:开源科研助手迈入“真智能”时代!
AI蛋白质设计零门槛!Biomni与Tamarind Bio强强联手,傻瓜式预测实现!
Biomni 正式开源:通用生物医药 AI 智能体登场,支持自然语言科研执行

测评方法


本次测评基于五个真实 notebook 执行样本,覆盖计算生物学的主要任务类型,逐一拆解 Biomni Lab 在各场景下的实际表现。

https://phylo.bio/use-cases
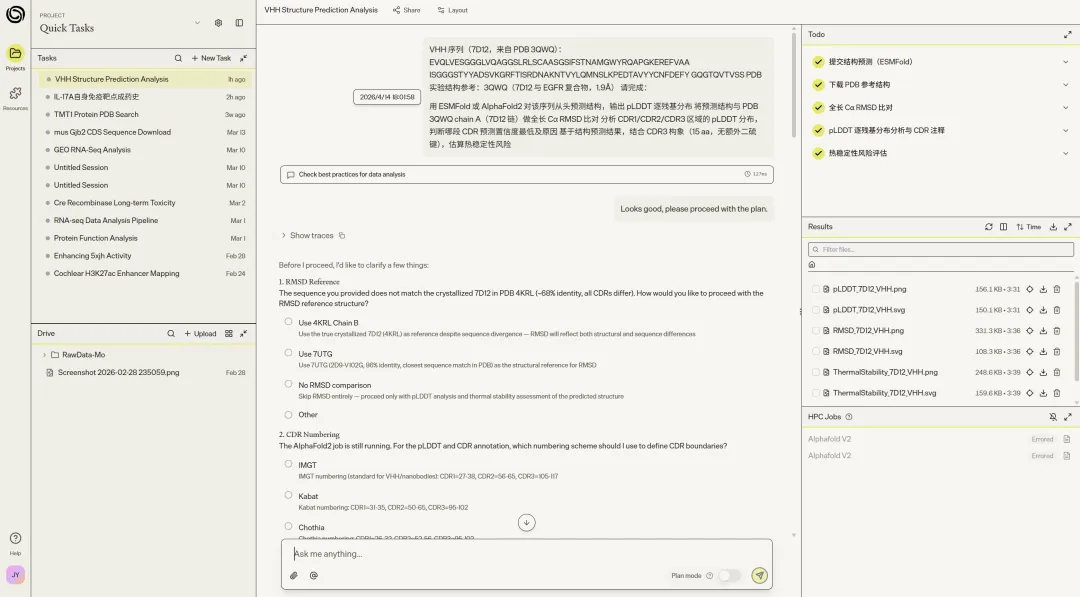
蛋白质结构预测(B2M AlphaFold)


平台调用 HPC 后端提交 AlphaFold2 作业,自动轮询结果,下载输出文件后解析 ranking_debug.json 和各模型的 pLDDT 打分,


最终生成包含置信度色带标注和多模型均值对比的发表级双图(PNG + SVG)。
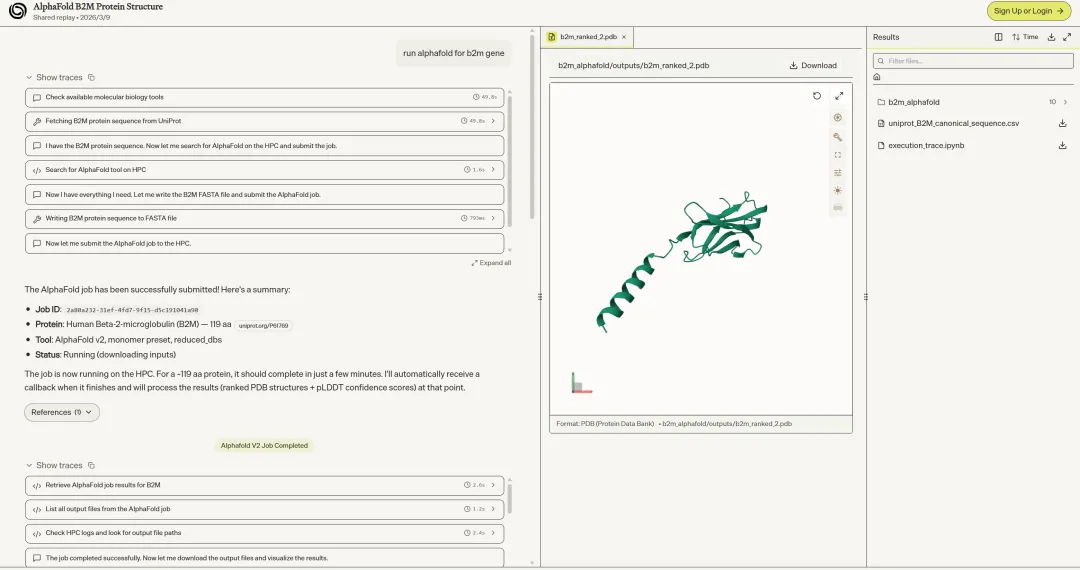
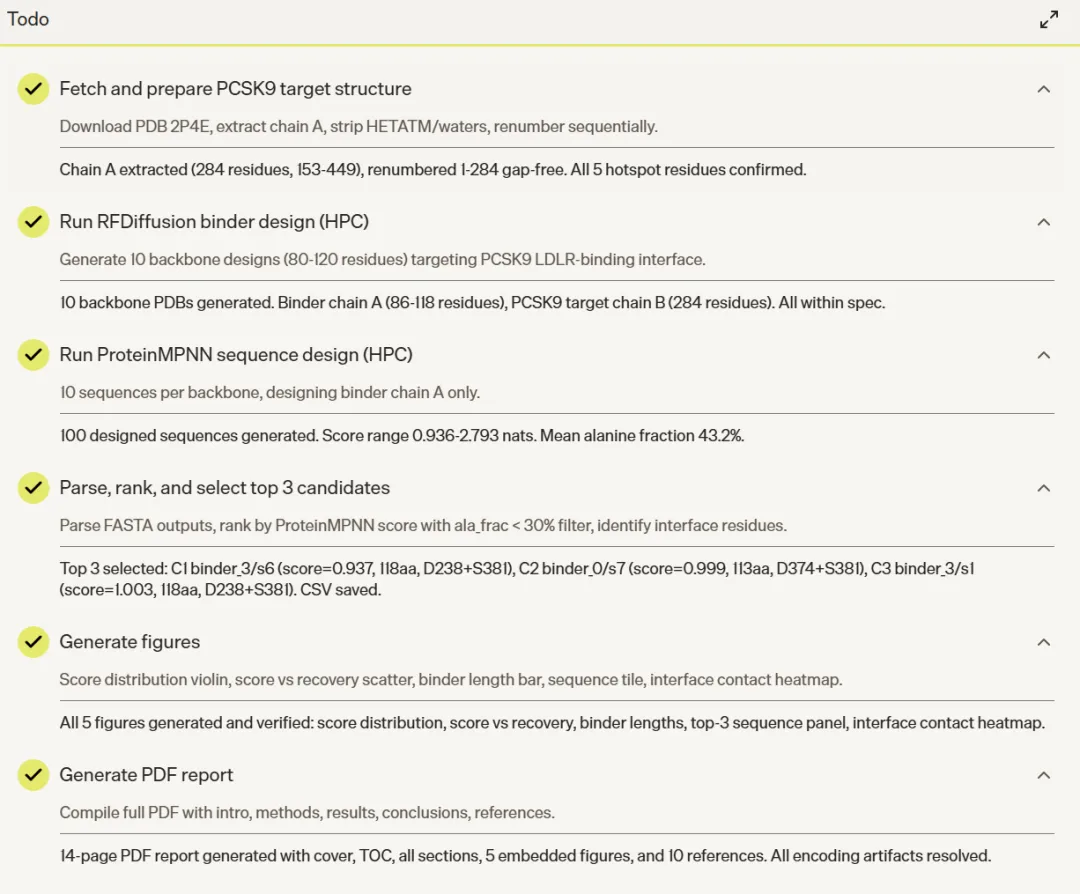
亮点在于全流程自动化——从作业提交到结果可视化,代码无需人工干预。五个预测模型的平均 pLDDT 集中在 92-94 区间,置信度分解逻辑(Very high / Confident / Low / Very low)完全遵循 AlphaFold 官方标准。HPC 结果检索 API 的设计也相当干净,hpc_api_get_results(job_id, output_dir) 一行调用即可。
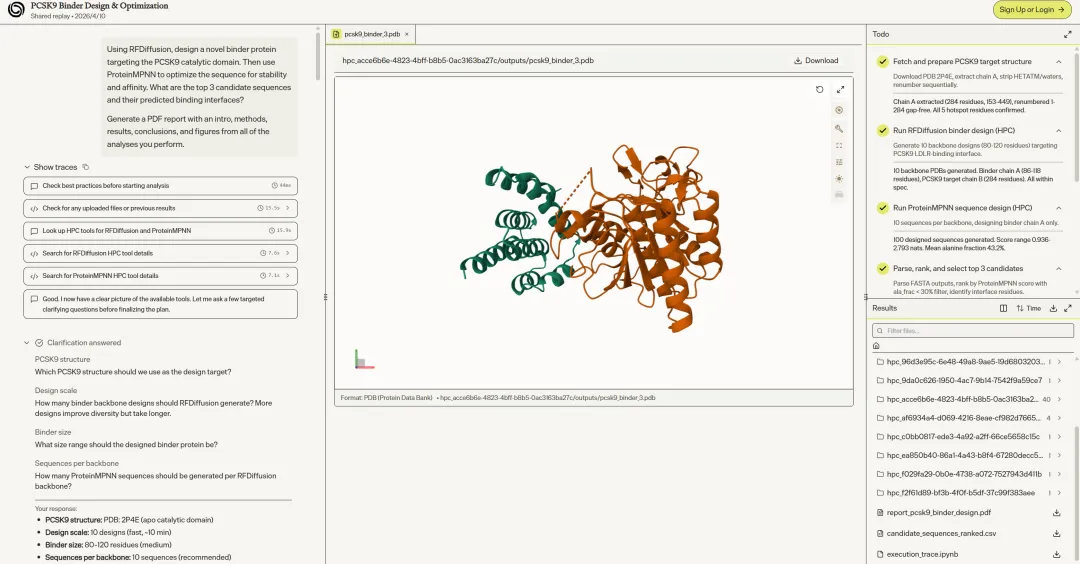
蛋白结合剂设计(PCSK9 Binder)


调用双工具链:先用 hpc.hpc_search_tools() 检索 RFDiffusion 和 ProteinMPNN 的参数规格,再下载 PDB 2P4E,提取 Chain A,验证 PCSK9 的五个热点残基(D238、R357、E366、D374、S381),排查 HETATM 记录和链间隙,最后生成适配 RFDiffusion 的 contig 配置。
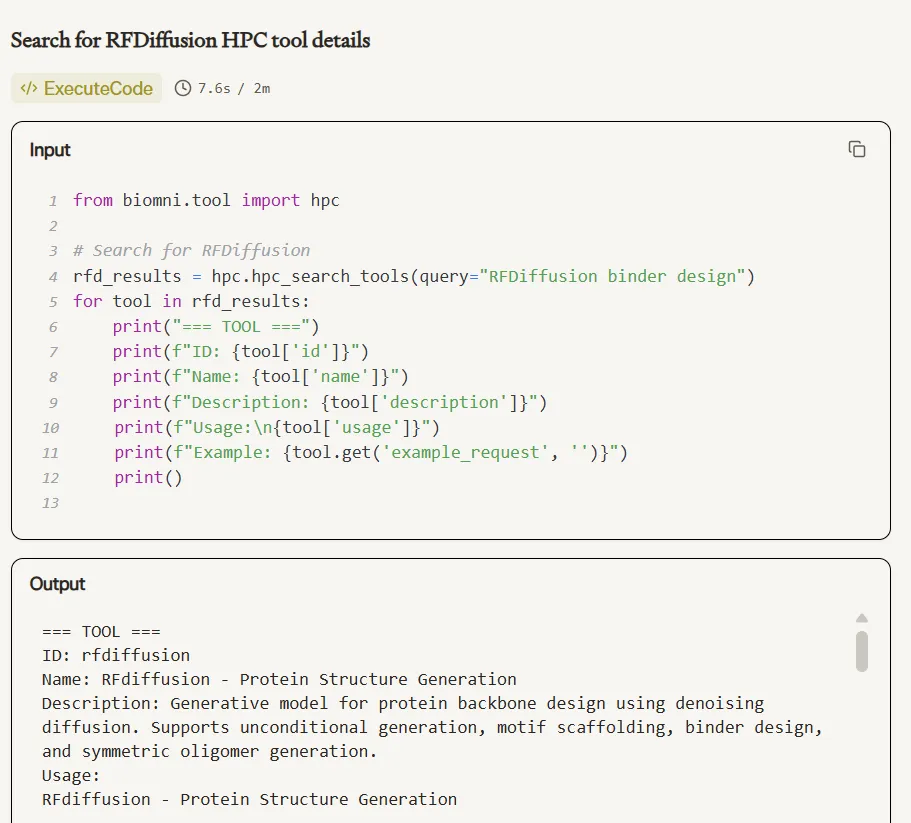
整个 PDB 预处理逻辑写得相当严谨——用 Biopython 精确过滤备用构象(altloc != A),并对热点残基逐一做 B-factor 检查以评估表面暴露程度。

工具检索接口抽象合理,科研人员可以通过语义查询发现平台支持的分析能力,而不必记忆固定 API 名称。
引物与克隆设计(Prime 编辑器)


任务难度较高——给定一段 3.7 kb 的 CDS 序列,找到内部 EcoRI 位点,穷举六个位置上所有可能的单碱基同义突变,筛选出不改变蛋白序列同时彻底消除酶切位点的方案,再设计带 BamHI / Kozak / FLAG tag 悬挂端的克隆引物。

Biomni Lab 的处理过程体现了良好的分子生物学知识深度:框内偏移量计算准确,同义突变遍历逻辑完整,最终选取 GGA→GGC(Gly,密码子偏好中性)作为最保守的修改策略。
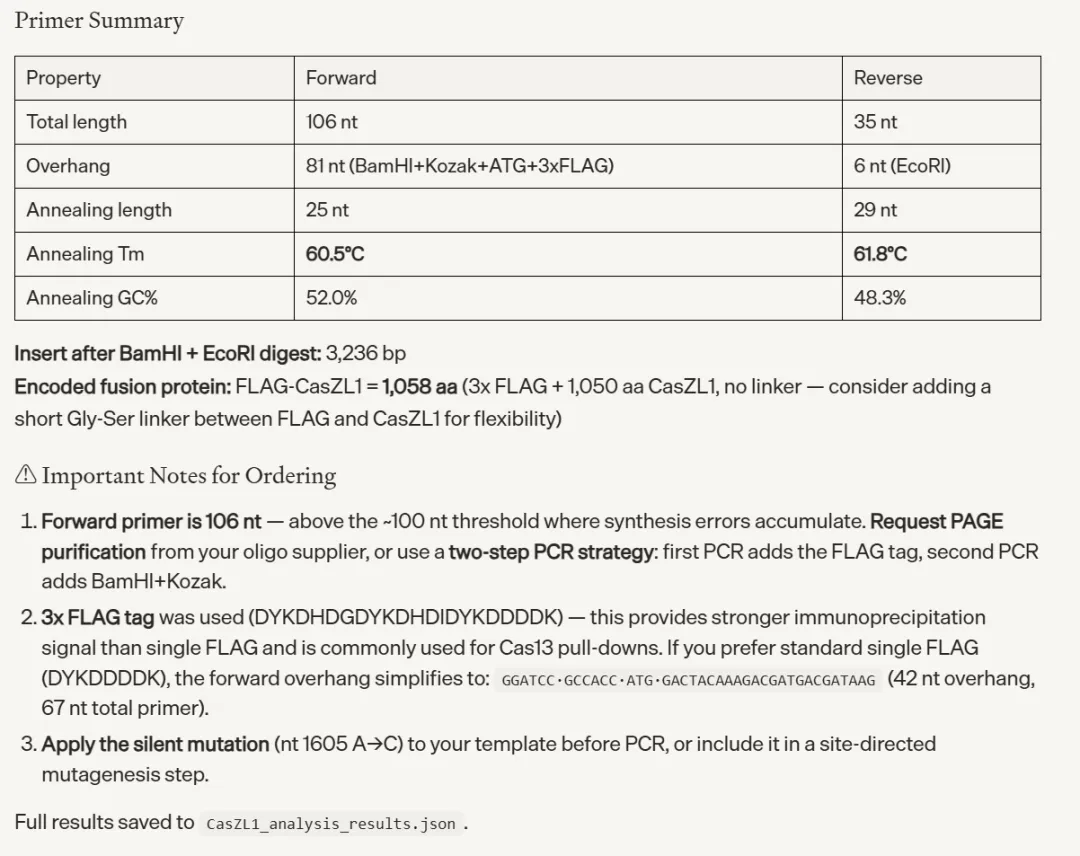
molecular_biology.design_pcr_primers_with_overhangs()工具接口将实验设计意图直接映射为函数参数,对湿实验科学家友好。

小分子活性数据
挖掘与模型训练(PCSK9 ChEMBL)


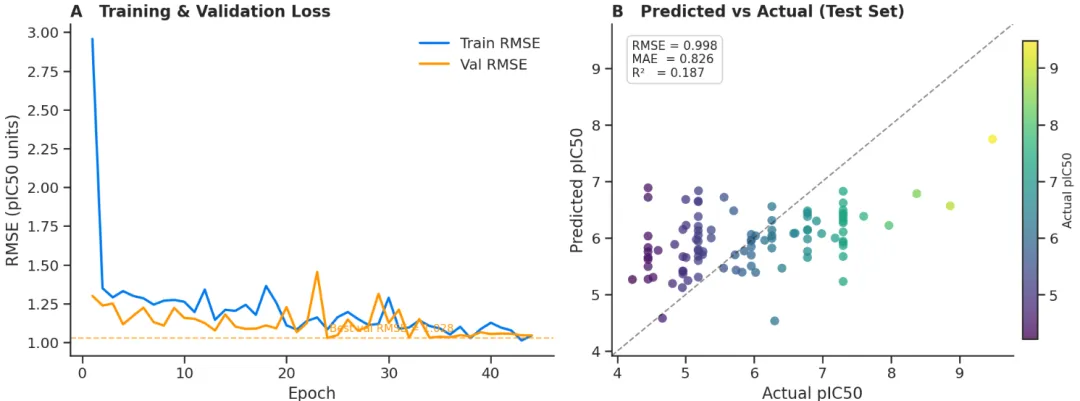
这是五个任务里流程最完整、技术深度最高的一个。Agent 从零搭建了一条端到端的 AI 药物发现 pipeline:从 ChEMBL 批量获取 PCSK9 的 IC50、Ki、Kd 数据,经过单位标准化、关系过滤(只保留精确测量值)、数据质量标记清洗、RDKit SMILES 验证、按 pIC50 四分位数分层划分训练/验证/测试集,再将每个分子转换为带 21 维原子特征和 6 维键特征的分子图,训练一个带边特征的 MPNN 模型,加入 BatchNorm、Dropout、梯度裁剪和早停机制,最终在测试集上输出 RMSE、MAE、R² 指标及预测散点图。
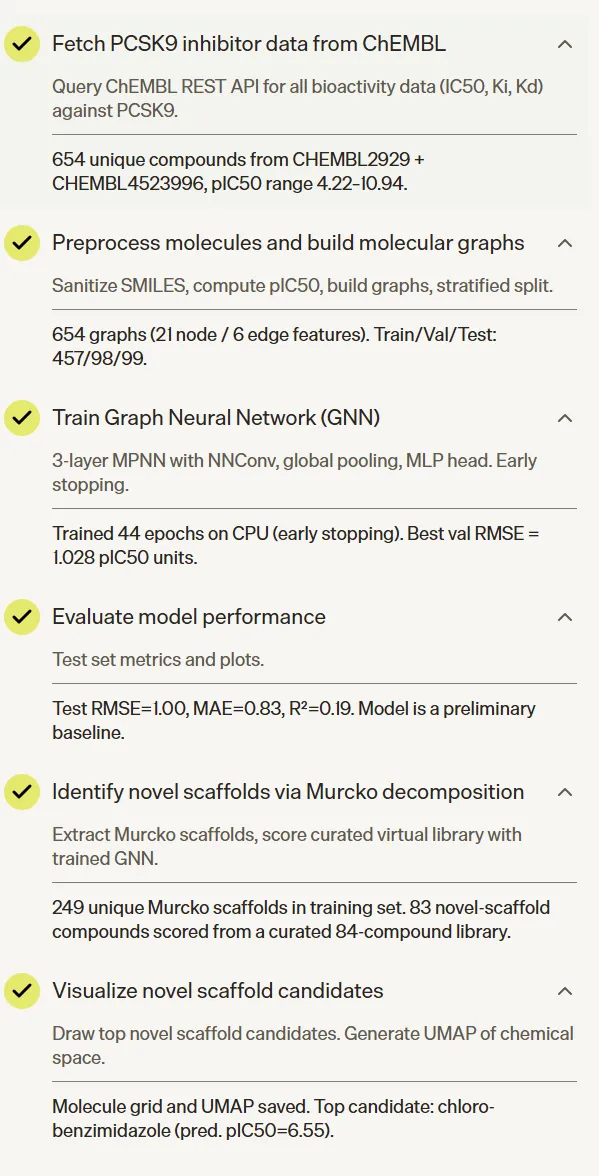
模型训练完成后,Agent 并未停在评估环节,而是继续提取训练集 Murcko scaffold,构建一批药物样小分子虚拟库,过滤掉与训练集重叠的骨架,用训练好的 GNN 对新骨架候选分子打分排序,最后计算 Morgan 指纹并用 UMAP 将已知 PCSK9 抑制剂和新骨架候选分子投影到同一化学空间,高亮 Top 5 候选。
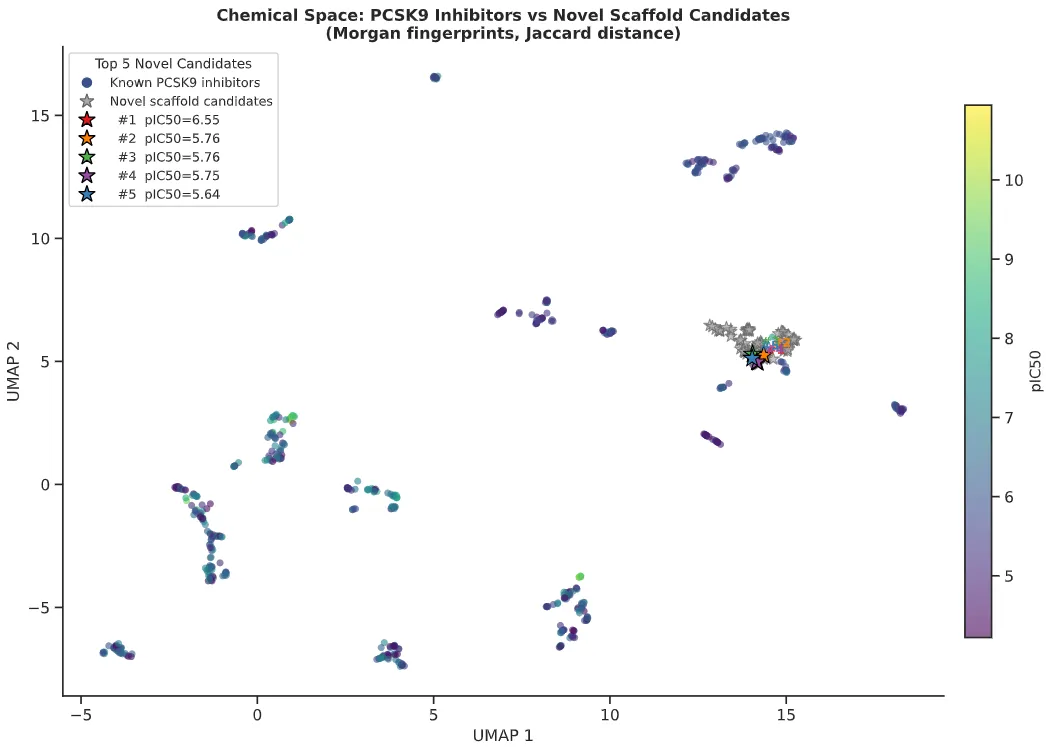
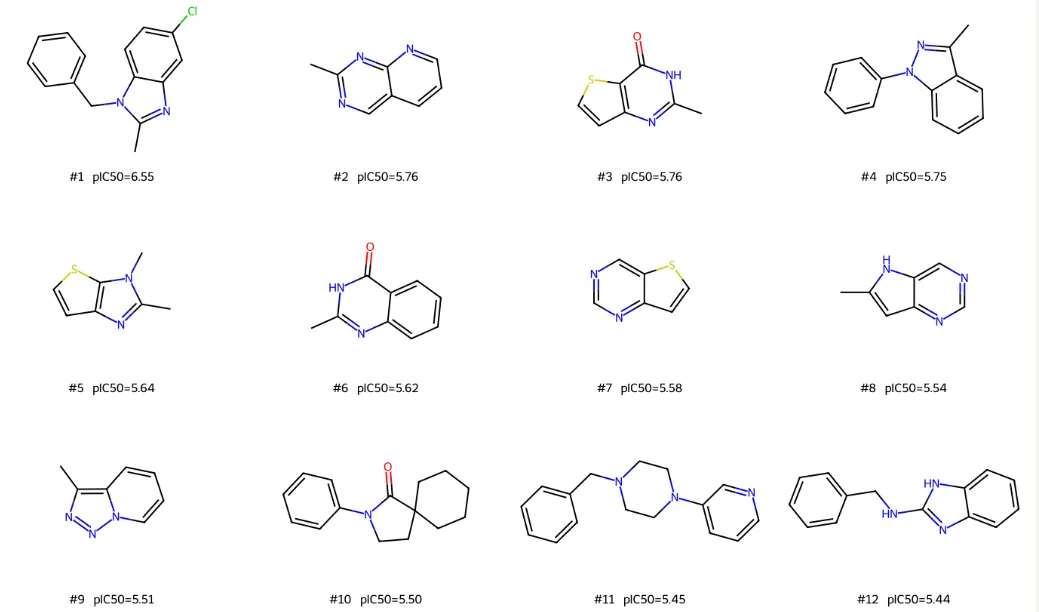
整个流程覆盖了从数据获取、模型训练到虚拟筛选的完整药物发现闭环,且每个环节的工程细节处理得相当规范,输出图表达到直接写入论文的质量。
差异表达分析
(MASH DEG,DESeq2)


这个任务的完成度在五个 demo 里最高,最终输出不是一个 notebook,而是一份完整的 PDF 科研报告。

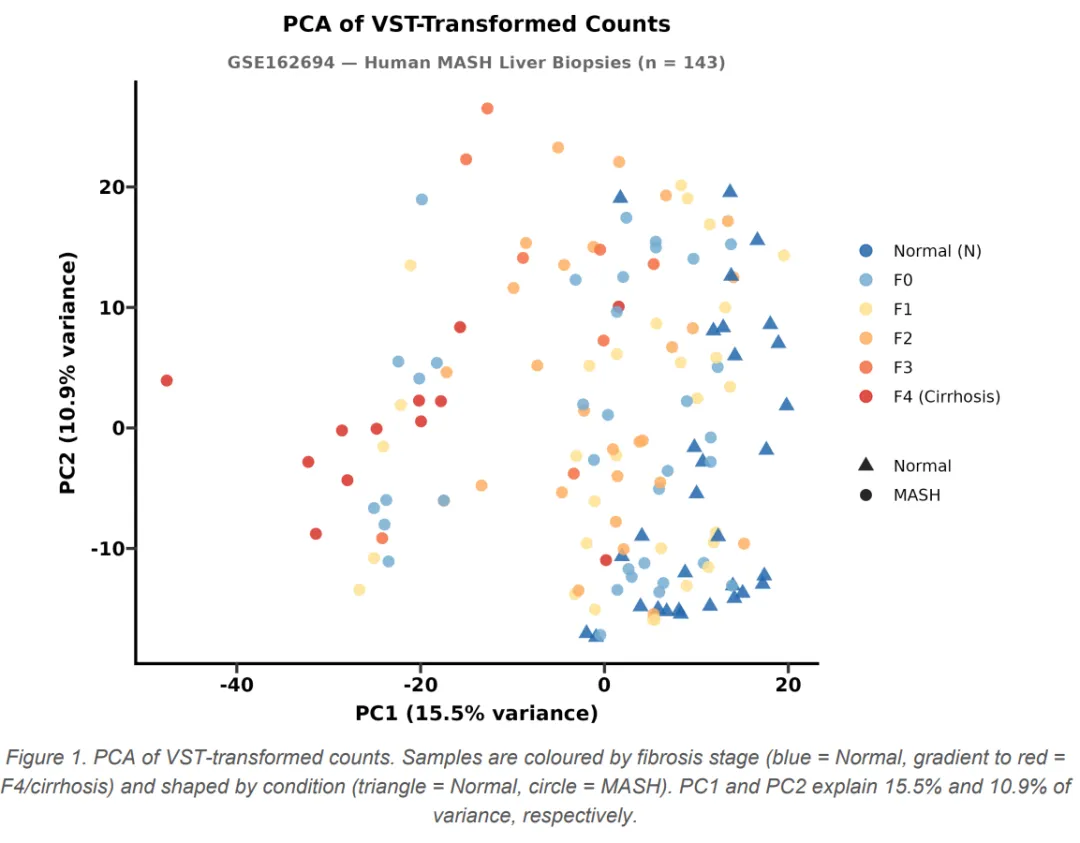
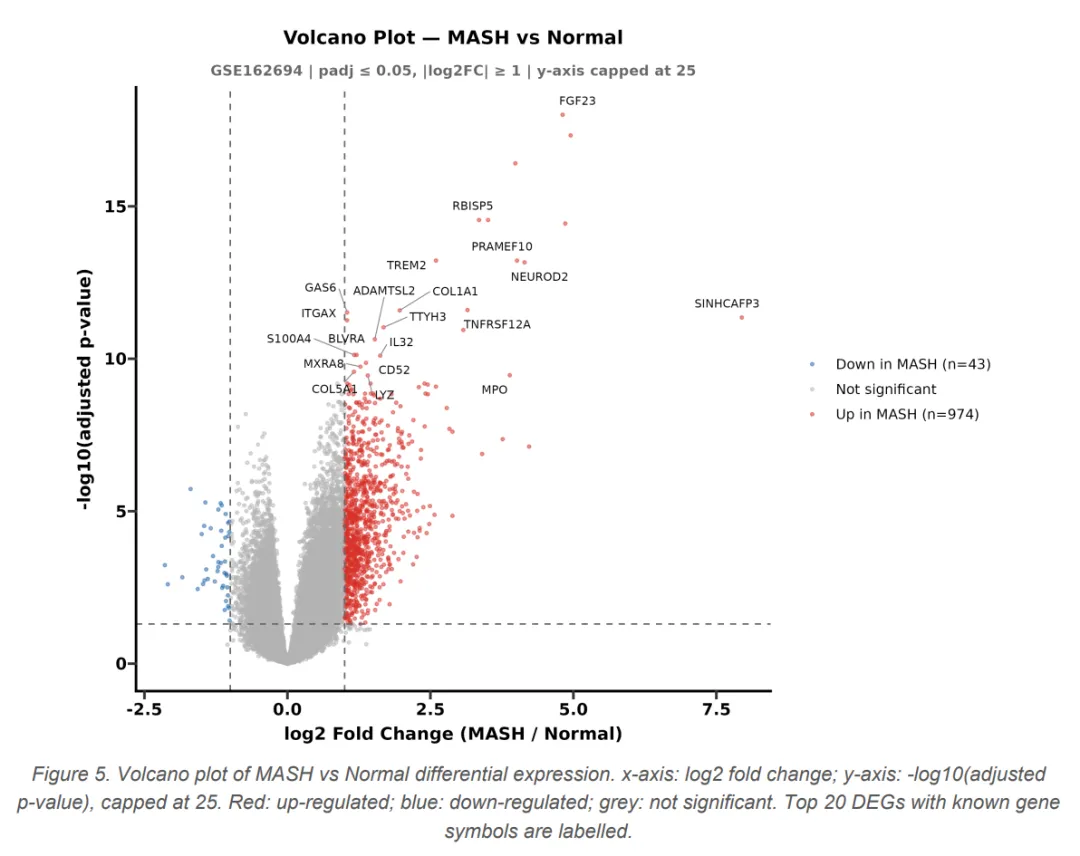
流程从 GEO 下载 GSE162694 数据集(143 例人类 MASH 肝活检 + 31 例正常肝组织的 RNA-seq 原始计数)开始,解析 SOFT 格式元数据提取纤维化分期(F0-F4 及 Normal),验证样本 ID 完全匹配后进入 R 环境。DESeq2 全流程走完——低表达基因过滤、构建 DESeqDataSet、负二项分布 Wald 检验、apeglm LFC 收缩,最终在 padj ≤ 0.05 且 |log2FC| ≥ 1 的阈值下识别出 1017 个显著 DEG(974 个上调、43 个下调)。

图表输出覆盖 VST-PCA图、离散度估计图、MA 图、火山图、Top50 DEG 热图(带纤维化分期列注释)、GO BP 富集点图和 KEGG 通路点图,全部以 300 dpi PNG 保存。
最后一步尤其值得注意:Agent 用 ReportLab 从零构建了一份结构完整的 A4 PDF 报告,包含封面、引言、方法、结果、结论、局限性、未来方向和参考文献八个章节。

正文引用了 DESeq2、apeglm、clusterProfiler 的原始文献,样本构成以格式化表格呈现,所有图表以图注嵌入对应章节。这份报告的格式和内容完整度,已接近可以直接投稿的补充材料水准。


综合评估


五个任务覆盖了结构预测、蛋白设计、分子克隆、AI 药物发现和转录组分析五个方向,共同呈现出几个一致的特点。
工具调用的抽象层次做得比通用 coding agent 更贴合科研习惯,无论是 HPC 作业调度、语义工具检索还是专有生物信息学接口,调用方式对科学家友好。图表输出质量统一达到发表标准,省去大量后处理工作。任务完成度普遍较高,尤其 GNN 虚拟筛选和 DESeq2 报告两个任务,端到端的完整性明显超出同类工具的常规水平。
需要说明的是,示例 notebook 是经过筛选的成功案例,无法通过这些材料评估平台在陌生任务、错误输入或边界条件下的表现。对于想真正评估平台能力的用户,建议用自己实际遇到的问题做测试。
如果你想跟进这类“AI × 生物资讯”的最新进展,欢迎关注

