 夜雨聆风
夜雨聆风点击左下角 阅读原文 查看论文原文
一篇 LNP × AI 论文,应该怎么读?——从构象、表征到模型的完整拆解(含读图方法)
LNP × AI/ML 系列推文
这两年以来,LNP 领域当中关于 AI/ML 的工作数量明显在增加。
模型变得越来越复杂,指标也变得越来越好看:
R²从 0.6 提升到了 0.8,Spearman 相关性从 0.7 提升到了 0.87。
但如果你认真读过几篇之后,很可能会有一个不太舒服的感觉:
这些模型看起来显得很"聪明",但你却更不理解 LNP 了。
它们可以来进行结果的预测,却很少去解释原因。
预测在进步,理解在原地。
这篇来自 Nature Biomedical Engineering 的工作:
Artificial intelligence-guided design of LNPs for in vivo targeted mRNA delivery via analysis of the spatial conformation of ionizable lipids
提供了一种不太一样的路径:
从分子构象出发,再来引入 AI,而不是直接去做黑箱预测。
这篇文章本身值得去读,但更重要的是:
它可以作为一个模板——来教我们如何真正读懂一篇 LNP × AI 论文。
下面按照"实际读论文的顺序",一步一步来拆解。
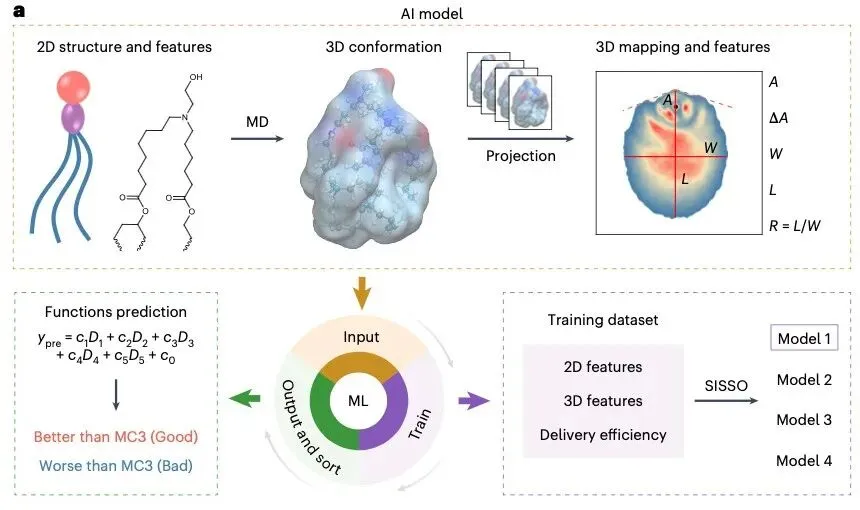
四个概念
在开始读图之前,我们需要先建立四个关键概念的认知框架。这四个概念贯穿整篇论文,理解它们,你才能真正读懂这篇 LNP × AI 论文。
第一个概念:特征(feature)
特征是机器学习模型用来进行预测的输入变量,是模型对原始数据的数学抽象。
特征是模型唯一看到的东西。模型不认识分子,不认识结构,它只认识数字。这 28 个数字就是模型眼中的"脂质"。
角度(A):脂质分子的弯曲角度
长度(L):分子的最长维度
宽度(W):分子的最宽维度
比值(L/W):长度与宽度的比例
如果特征没有包含关键信息,模型永远学不到东西。这篇论文选择 28 个特征,而不是 100 个或 1000 个,是因为这 28 个特征足够描述构象分布的核心信息,同时又避免了过拟合。
第二个概念:表示(representation)
表示是数据在机器学习系统中的编码方式,同一对象可以有不同的表示形式。
同一个分子,可以有不同"翻译"方式。机器学习不理解分子本身,它只理解你给它的"翻译版本"。如果你给的"翻译"丢掉了关键信息,模型再强也没用。
SMILES 字符串:用文本表示分子结构(如"CCO"表示乙醇)
分子描述符:用物理化学参数表示(如分子量、logP)
图结构:用节点和边表示原子和化学键
构象密度图:用 3D 空间概率分布表示(本文创新)
这篇论文的核心创新,就是把脂质的表示方式从"化学结构"升级为"构象分布"。之前的研究用 SMILES 或描述符,丢掉了构象信息;这篇论文用构象密度图,保留了关键的空间信息。
第三个概念:过拟合(overfitting)
过拟合是指模型在训练数据上表现很好,但在新数据上表现很差的现象,本质是模型记住了训练数据的噪声而非规律。
过拟合就是"死记硬背"。模型把训练数据的细节都记住了,但没学会真正的规律。在 LNP 领域非常常见:同一个实验室做出来的模型很好,换一个实验室就失效。
如果用 1000 个特征训练 1408 个脂质,模型可能记住每个脂质的"样子"
但遇到新脂质时,它就完全不会预测了
这就是为什么作者选择 28 个特征:既要保留关键信息,又要避免过拟合
判断方法是看外部验证,看分布外数据。如果模型只能预测训练集内的数据,那它可能过拟合了。
第四个概念:分布外数据(Out-of-Distribution, OOD)
分布外数据是指与训练数据来自不同分布的测试数据,用于评估模型的泛化能力。
分布外数据就是"模型没见过的数据类型"。如果模型能预测这些"没见过"的数据,说明它学到了普适规律;否则,它只是"记住了训练数据"。
在这篇论文中
训练数据是作者合成的 1408 种脂质
分布外数据是MC3、SM-102、ALC-0315(商业脂质)
模型能预测 MC3 和 SM-102 的递送效率,说明学到了规律
如果模型能预测分布外数据,才说明它学到了"规律"。不能预测分布外数据 → 模型基本不可用。这篇论文的模型能预测商业脂质,证明了它的泛化能力。
四个概念的关系
特征(模型看到什么?) → 表示(如何编码?) → 过拟合(是否死记?) → 分布外数据(能否泛化?)
理解这四个概念,才能方便读懂这篇论文的创新点:
特征选择:28 个构象特征,足够描述关键信息
表示创新:从化学结构升级到构象分布
避免过拟合:用 SISSO 线性模型,而不是深度学习
验证泛化:能预测 MC3、SM-102 等分布外数据
一、先不要去看模型:它在研究哪一层变量?
如果只看引言部分,会看到一些熟悉的变量:
亲水头基(head)、疏水尾部(tail)、连接子(linker)的结构调控,pKa 值、不饱和度、支链结构,蛋白冠(protein corona)与器官靶向。
这些在 LNP 文献当中都很常见,都属于 结构层。
但这篇文章真正往前推进的一步是:
把"构象(conformation)"单独作为变量层来引入。
为什么这一步会很重要?
因为在真实体系当中:
分子在发生动态变化,环境在发生变化(从有机相到水相),电荷状态也在发生变化(内体酸化)。
真实情况更接近于:
一个结构 → 多种构象 → 多种功能路径
而不是:
一个结构 → 一个功能
这是读这类论文的第一个判断:
它在研究哪一层变量?
组分(composition)、工艺(process)、结构(structure)、构象(conformation)、生物学(biology)。
很多机器学习工作停留在结构层,这篇往前推进了一层。
如果变量层选错了,后面的模型再复杂也只是在错误的问题上去做优化。
二、Figure 1:整篇论文的"说明书"
Figure 1a = 方法 + 数据 + 逻辑的压缩表达。
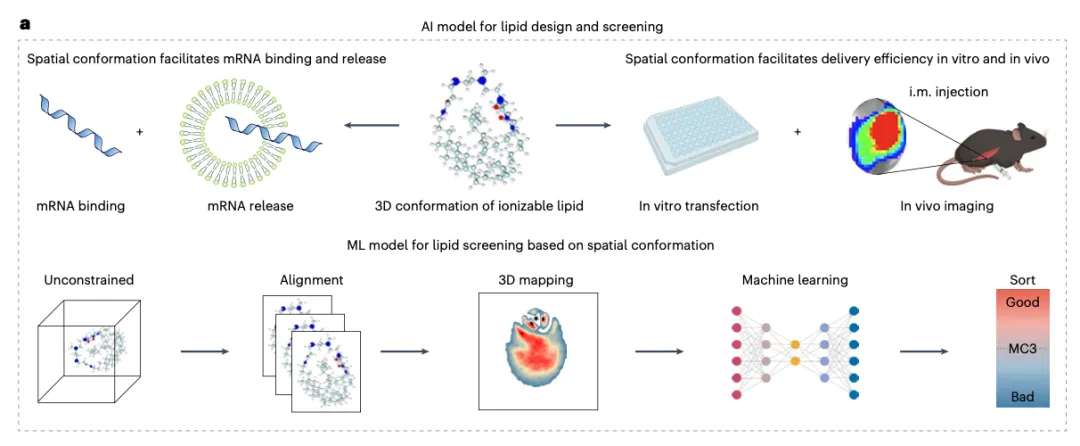
正确的读法不是"去看图",而是要去问 3 个问题:
第一个问题:输入是什么?
这篇不是制剂配方,不是分子描述符,而是 分子动力学(MD)模拟得到的构象分布。
关键理解:AI 的输入不是"分子",而是"构象数据"。
如果输入层没有去包含关键信息,后面的模型再强也学不到东西。
第二个问题:中间做了什么变换?
你会看到三个关键步骤:对齐(alignment)、投影(projection)、特征提取(feature extraction)。
本质是:
物理世界(分子在动)→ 标准化(对齐)→ 降维(投影)→ 数字表示(特征)
一句话来总结:
机器学习学的不是结构,而是"表示方式(representation)"。
第三个问题:输出是什么?
mRNA 递送效率、器官靶向性。
Figure 1 的一句话读法:
无约束构象 → 对齐 → 3D 映射 → 机器学习 → 排序
AI 只发生在这条链的最后一步,前面所有步骤都在决定 AI 能够看到什么。
📌 一个可以直接复用的读图方法
以后去读任何 LNP × AI 论文里的"方法图",都可以去用这四步:
看输入(Input):模型到底"看到了什么"?
看变换(Transformation):信息是如何被压缩或转换的?
看表示(Representation):最终变成了什么形式(特征或嵌入向量)?
看输出(Output):模型预测的到底是什么?
任何 AI 模型,本质都是:输入 → 表示 → 输出
模型从不理解分子,它只理解你给它的表示。
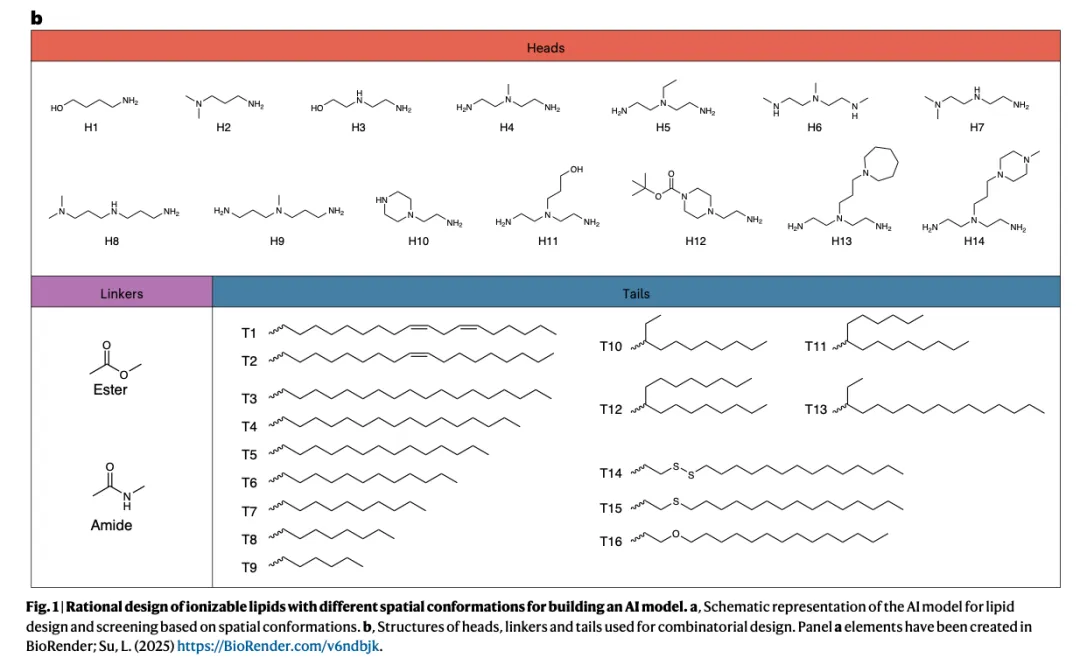
Figure 1b:脂质库的设计策略(组合化学)
Figure 1b 展示了作者是如何设计这一千四百零八种脂质的。
这部分的读法是:
不是去记每个结构,而是理解"组合逻辑"。
作者用了组合化学的思路:
头基(Heads):十四种(H1 到 H14),不同数量的氨基、不同取代方式。
连接子(Linkers):两种,酯键(Ester)和酰胺键(Amide)。
尾部(Tails):十六种(T1 到 T16),不同链长、不同不饱和度、不同杂原子。
这个设计的关键在于:
不是随机组合,而是"有方向地覆盖化学空间"。
改变头基 → 去看电荷分布的变化。
改变尾部 → 去看堆积和相行为的变化。
改变连接子 → 去看稳定性和靶向性的变化。
为什么这一点很重要?
因为模型之后能不能"学到东西",取决于你有没有提供"结构变化的梯度"。
如果训练数据只是随机组合,模型学到的只是"统计相关性"。
但如果有方向地去覆盖化学空间,模型有机会学到"结构 - 功能关系"。
三、Figure 2:最容易"看不懂"的图
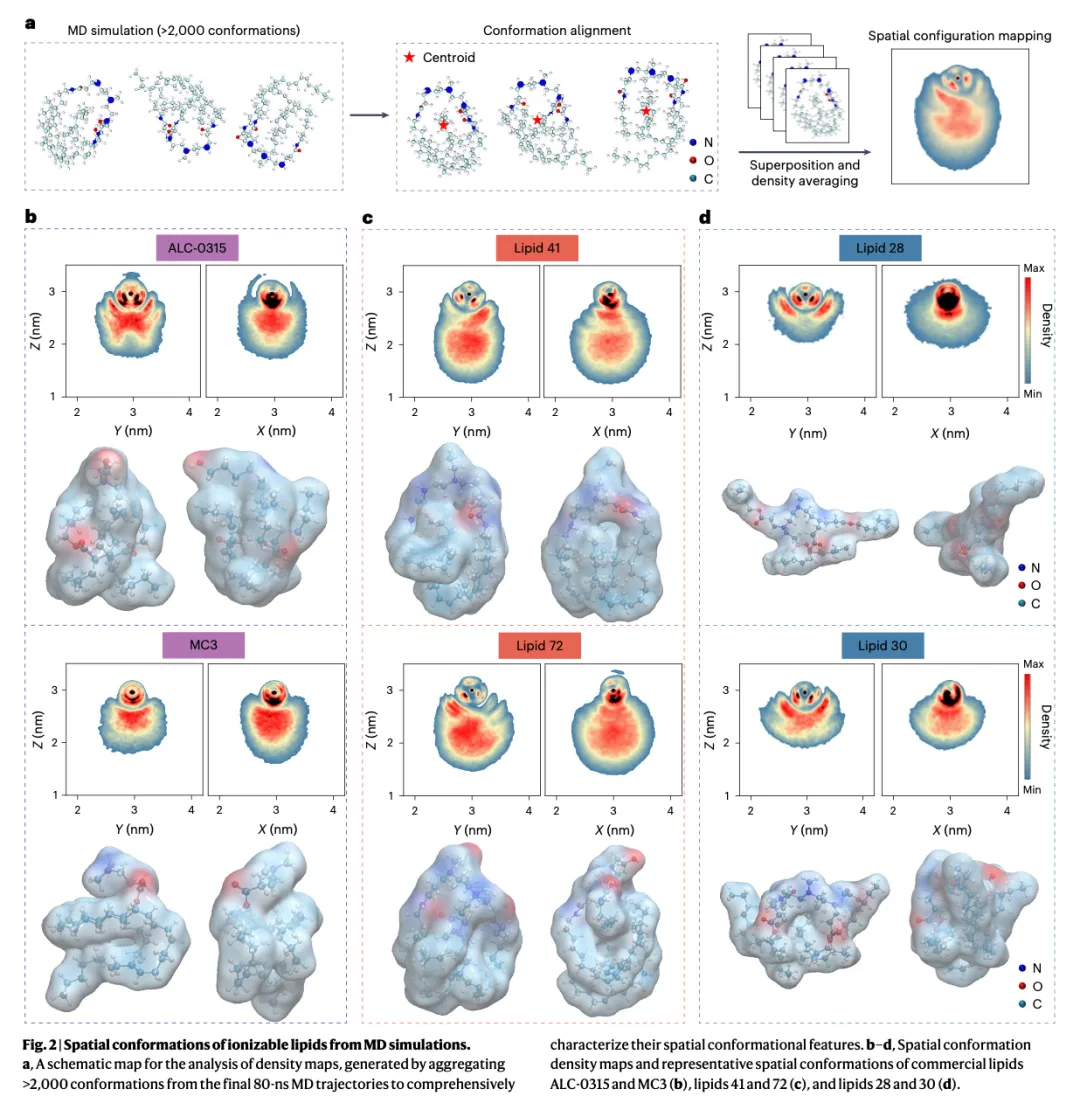
先来讲解一个核心概念:
什么是"构象密度图"?
可以这样去理解:
一个分子在不断运动 → 记录每一帧的位置 → 叠加 → 得到"出现概率"
所以这张图表达的不是:"分子长什么样"
而是"分子更倾向于怎样折叠"
图里每个元素代表什么?
红色区域代表原子出现概率高(构象稳定区),蓝色区域代表原子出现概率低(构象灵活区)。
本质是一个概率分布图(probability distribution)。
为什么这张图很关键?
因为它把一个隐变量变成了可见变量:
构象分布 ≠ 单一结构。
如果只去看单一结构,会错过最关键的信息:
不同脂质不只是结构不同,而是"构象分布不同"。
这一步是整篇论文的基础,如果没有这一步,后面的机器学习就没有意义。
正确理解 Figure 2 的方式:
这不是"结构图",而是"分布图"。
红色区域 = 分子最常出现的构象,蓝色区域 = 分子很少出现的构象。
当你看到这个图时,应该去问:
这个脂质的构象分布是"集中"还是"分散"?
是"锥形"还是"球形"?
头部是"暴露"还是"被遮挡"?
这些问题比"结构是什么"更重要。
如何在 10 秒内判断一个脂质的构象类型?
拿到 Figure 2,不要整体去看,按照这个顺序:
先看"头部位置"
去问:头部是在外侧(暴露),还是被尾部包住(遮挡)?
判断意义:暴露更可能参与 mRNA 结合,遮挡则结合受限。
再看"整体形状"
去问:是"锥形(cone-shaped)",还是"接近球形或折叠"?
判断意义:锥形有利于膜扰动(内体逃逸),球形更稳定但不活跃。
看"分布是否集中"
去问:红色区域是否集中在一个区域?还是分散?
判断意义:集中说明构象稳定(单一行为),分散说明多构象(行为不确定)。
最后再看"细节参数"
这一步才是 AI 用的特征(角度、长度、宽度、比值)。
Figure 2 的三类典型构象(结合论文脂质编号)

紧凑锥形构象(Cone-shaped, Compact)
代表脂质:ALC-0315、MC3、Lipid 72
构象特征:头部(氨基)暴露在外侧,尾部聚集在另一侧,整体呈现明显的"头大尾小"锥形,密度图红色区域集中。
论文意义:这类构象有利于 mRNA 结合(头部暴露)和内体逃逸(锥形促进膜融合)。
伸展构象(Extended)
代表脂质:Lipid 41
构象特征:分子链更加伸展,头部和尾部分布更分散,密度图红色区域相对分散,整体形状更接近椭球形。
论文意义:这类构象可能稳定性更好,但 mRNA 结合能力可能较弱。
折叠/不规则构象(Folded/Irregular)
代表脂质:Lipid 28、Lipid 30
构象特征:分子链发生折叠,头部可能被尾部遮挡,密度图红色区域分散且不规则,整体形状无明显对称性。
论文意义:这类构象可能 mRNA 结合受限(头部被遮挡),递送效率较低。
不是"结构决定功能",而是"构象分布决定功能"
同一化学结构的脂质,在不同环境下会呈现不同构象。
论文通过 MD 模拟展示了:
每个脂质有 >2,000 个构象,这些构象被对齐、叠加、平均,最终得到构象密度图。
这才是 AI 模型真正学习的东西:不是化学结构,而是构象分布的表示。
Figure 1b 设计了一千四百零八种脂质(不同头基、连接子、尾部组合)
Figure 2 展示了这些脂质的实际构象:
同样的化学结构 → 多种构象
不同化学结构 → 可能相似的构象分布
所以论文的创新点在于:
不是用化学结构预测功能,而是用构象分布预测功能。
这才是"从结构走向构象"的真正含义。
四、Figure 3:构象 - 功能关系验证(核心机制)
这是整篇论文的功能验证核心图,回答了最关键的问题:
"构象分布的差异,是否真的会导致递送效率的差异?"
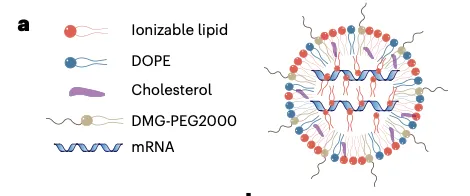
Figure 3a:LNP 配方示意图
展示了 LNP 的四组分结构:可电离脂质(红色)、DOPE(蓝色)、胆固醇(紫色)、DMG-PEG2000(黄色)、mRNA(波浪线)。
所有脂质都用相同的四组分配方,唯一的变量是可电离脂质的结构。这样设计是为了排除配方的干扰,单独看脂质构象的影响。

Figure 3b:冷冻电镜(cryo-TEM)图像
展示了 Lipid P1 和 Lipid 11 的 LNP 形貌。
关键观察:两种脂质都形成了均匀的球形 LNP,粒径约 100nm 左右,内部结构清晰可见。
排除"形貌差异"这个干扰因素。如果 P1 和 P11 的递送效率不同,不是因为 LNP 形貌不同,而是构象不同。

Figure 3c:热图(Heat map)- 核心数据
展示了 96 种脂质的体外转染效率。
读图方法:横轴是脂质编号(1-96),纵轴是不同脂质(按头基/尾部/连接子分组),颜色是发光强度(log₁₀),越红效率越高。
关键发现:有些脂质效率很高(深红色,如 Lipid 1, 29, 58),有些脂质效率很低(白色,如 Lipid 9, 17)。同一头基的不同尾部,效率差异巨大。
证明脂质结构(进而构象)对递送效率有决定性影响。
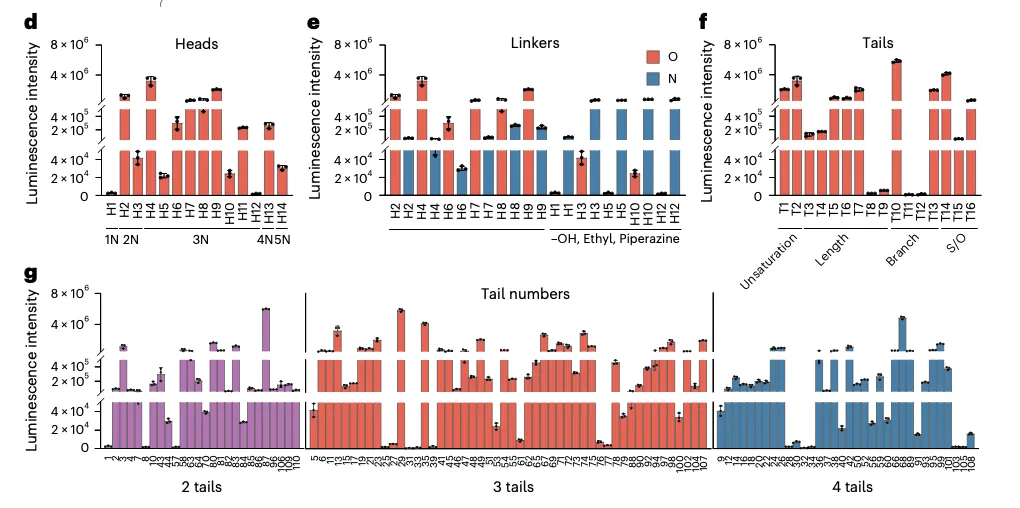
Figure 3d-g:构象特征与效率的关系(核心机制)
这部分是整篇论文的机制核心,把 Figure 2 的构象分类和 Figure 3c 的功能数据关联起来了。
3d 头基(Heads)的影响:横轴是不同头基类型(1N, 2N, 3N, 4N5N),纵轴是发光强度。结论:头基氨基数量影响效率,但不是唯一决定因素。
3e 连接子(Linkers)的影响:横轴是不同连接子(酯键 O / 酰胺键 N)。关键发现:酯键(红色)普遍优于酰胺键(蓝色)。论文解释:酯键更易降解,有利于 mRNA 释放。
3f 尾部(Tails)的影响:横轴是不同尾部结构(不饱和度、链长、支链、杂原子)。关键发现:适度不饱和最优(T2, T3),链长 C14-C18 最优,适度支链优于直链,含硫/氧的尾部效率更高。
3g 尾部数量的影响:横轴是 2 尾 / 3 尾 / 4 尾。关键发现:3 尾脂质普遍优于 2 尾和 4 尾。论文解释:3 尾脂质更容易形成锥形构象(呼应 Figure 2)。
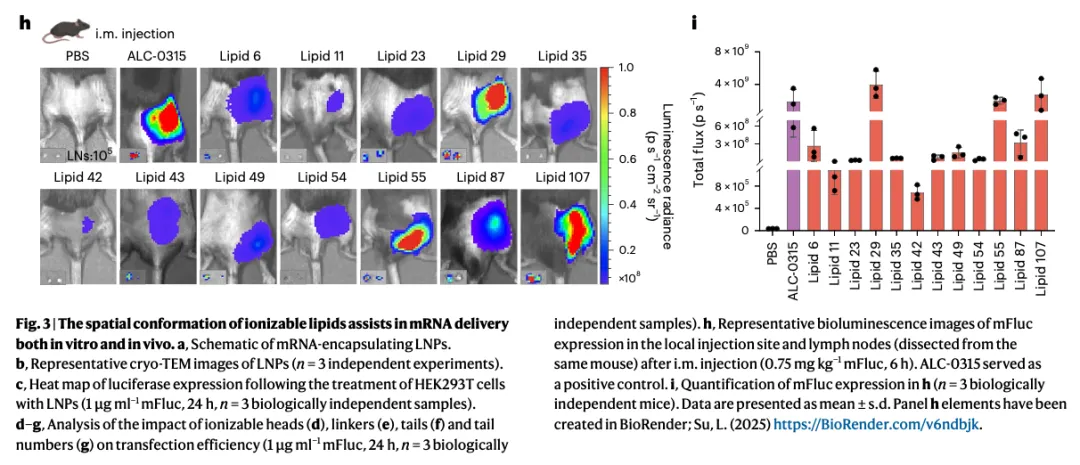
Figure 3h-i:体内实验验证
3h 活体成像图:展示了不同脂质 LNP 在小鼠体内的 mRNA 表达。关键观察:Lipid 29, 35, 87, 107 效率很高(红色/黄色区域大),Lipid 6, 11, 42 效率较低(蓝色区域),ALC-0315(阳性对照)效率中等。
3i 定量分析:纵轴是总发光通量。关键发现:Lipid 29, 35, 107 的体内效率显著高于 ALC-0315,有些脂质体外好但体内差(如 Lipid 1),体内 - 体外相关性不完全一致。
证明了构象优化的脂质在体内也有效,但体内环境更复杂,需要综合考虑。
Figure 3 的核心结论
构象 - 功能关系得到验证
Figure 2 定义的构象分类,在 Figure 3 得到了功能验证:
紧凑锥形(Lipid 29, 35, 72)→ 递送效率高
伸展构象(Lipid 41, 72)→ 递送效率中
折叠构象(Lipid 28, 30)→ 递送效率低
结论:锥形构象(头部暴露、3 尾)确实递送效率更高。
结构 - 构象 - 功能的完整链条
Figure 3 建立了完整的因果链条:
化学结构(头基/连接子/尾部)→ 空间构象(Figure 2 的密度图)→ 递送功能(Figure 3 的发光强度)
这是论文的核心创新:
之前的研究只关注"结构 → 功能",跳过了"构象"这个中间层。
这篇论文明确证明:结构通过构象影响功能。
设计规则的提炼
基于 Figure 3d-g,论文提炼出了可操作的设计规则:
高效脂质的特征:3 个尾部(形成锥形构象)、酯键连接子(易于降解)、适度不饱和尾部(增加膜融合能力)、头部暴露(利于 mRNA 结合)。
这些规则直接指导了后续 P1-P6 脂质的设计(Figure 4)。
五、从构象到特征:AI 真正工作的地方
这一部分通常一句带过,但其实是整篇核心。
他们做了什么?
从密度图提取角度(A)、长度(L)、宽度(W)、比值(L/W),共二十二个构象特征,再加六个化学特征,得到二十八维特征向量。
必须理解的一个事实:
模型从来没有"看到分子",它只看到二十八个数字。
关键问题:
这二十八个数字,是否保留了关键信息?
如果特征没有去包含构象分布的核心信息,模型永远学不到东西。
一个更直观的理解
你可以把这个过程理解为:
真实世界:一个脂质 → 会摆很多姿势AI 世界:一个脂质 → 二十八个数字
关键问题变成:
这二十八个数字,是否能区分:
锥形和折叠、暴露和遮挡、稳定和多变。
不能区分机制的特征,本质上是噪声。
这是读 AI 论文最重要的一步:
不是去看模型,而是去看特征(feature)。
去问自己这二十八个特征,是否足够描述构象分布?
有没有丢掉关键信息?
如果是你,你会怎么去定义特征?
论文中的 SISSO 公式(核心表达式)
作者使用的 SISSO 模型,最终得到了一个由五个集体项(D1, D2, D3, D4, D5)组成的公式:

ypre = c1×D1 + c2×D2 + c3×D3 + c4×D4 + c5×D5 + c0
ypre = log₁₀(E_pre),E_pre 是预测的递送效率
c0 到 c5 = 线性系数(通过交叉验证确定)
D1 到 D5 = 从 28 个特征中选出的 5 个最关键特征
这个公式的意义是什么?
它不是黑箱,而是一个可解释的线性模型。
每个 D 项代表一个构象特征(比如头部暴露程度、尾部长度、锥度等),系数 c 代表这个特征对递送效率的贡献大小。
正系数:这个特征越强,递送效率越高。
负系数:这个特征越强,递送效率越低。
为什么选择线性模型,而不是深度学习?
因为数据量有限(1,408 种脂质),深度学习容易过拟合。
SISSO 的优势在于:小数据友好(几百个样本就能训练),可解释(每个特征的贡献清晰可见),泛化能力强(能预测分布外数据)
MC3 作为基线
论文以商业脂质 MC3 作为基线(E_base = 10⁵):
预测效率 > MC3 → "好脂质"
预测效率 < MC3 → "差脂质"
这个分类直接指导了后续 P1-P6 脂质的设计。
在这篇中保留了部分构象分布信息,但仍然是降维表示。
这是一个权衡:特征太少会丢掉信息,特征太多会导致过拟合(overfitting)。
六、Figure 4:模型结果该怎么看?
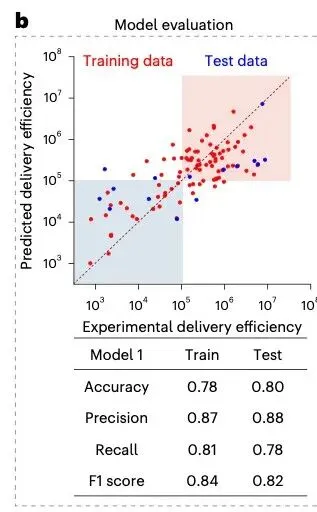
这是最容易被误读的部分。
常见误读方式:去看准确率,去看 R 平方,然后结束。
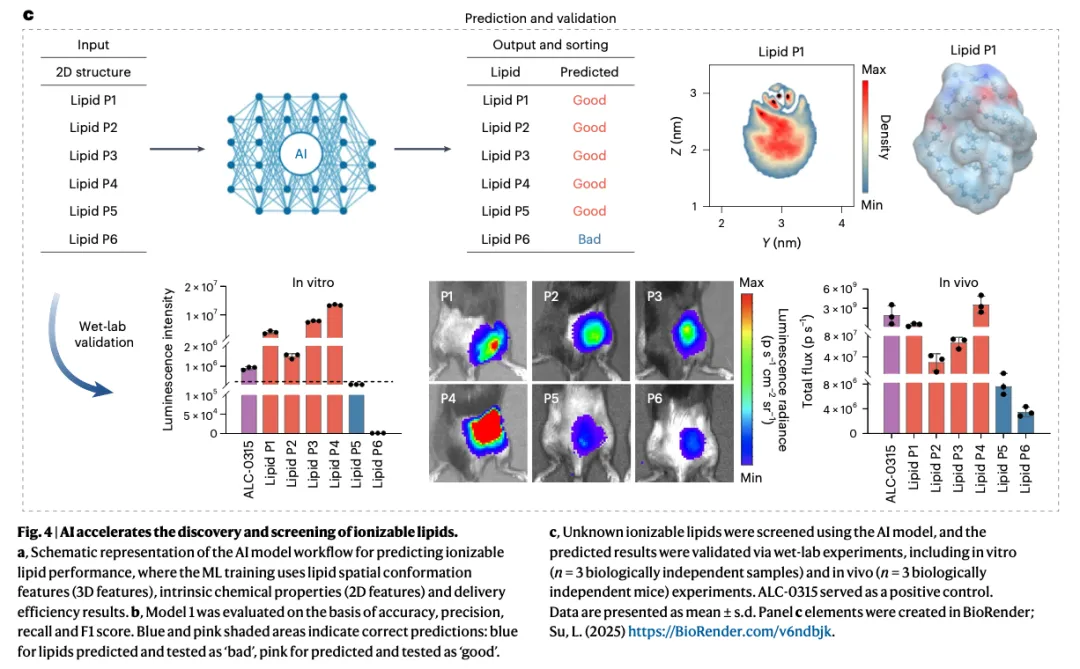
正确读法:去看三件事。
第一件事:有没有"训练和测试分离"?
如果没有,模型可能只是记住数据。
这篇论文多轮交叉验证,准确率约0.8。
这个结果是"合理"的,而不是"惊艳"的。
第二件事:有没有"分布外数据"?
这篇用了 MC3、SM-102、ALC-0315(商业脂质)。
这些不是训练数据分布的一部分。
这才是真正的泛化测试。
如果模型只能预测训练集内的数据,那它的价值有限。
但如果能预测分布外数据(如商业脂质),说明它真正学到了规律。
第三件事:有没有"新分子设计"?(最重要)
模型提出 P1 到 P6(新设计的脂质)。
实验结果:P1 到 P4 成功,P6 失败。
有失败,反而更可信。
如果模型说什么都好,那它可能只是"乐观估计器"。
但如果有成功有失败,说明它在真正预测。
Figure 4 的一句话总结:
模型的价值,不在拟合已知,而在预测未知。
七、Figure 5-8:应用验证(从机制到功能)
Figure 3 验证了构象 - 功能关系,Figure 4 展示了模型预测能力。
Figure 5-8 则进一步展示了从机制理解到实际应用的完整链条。
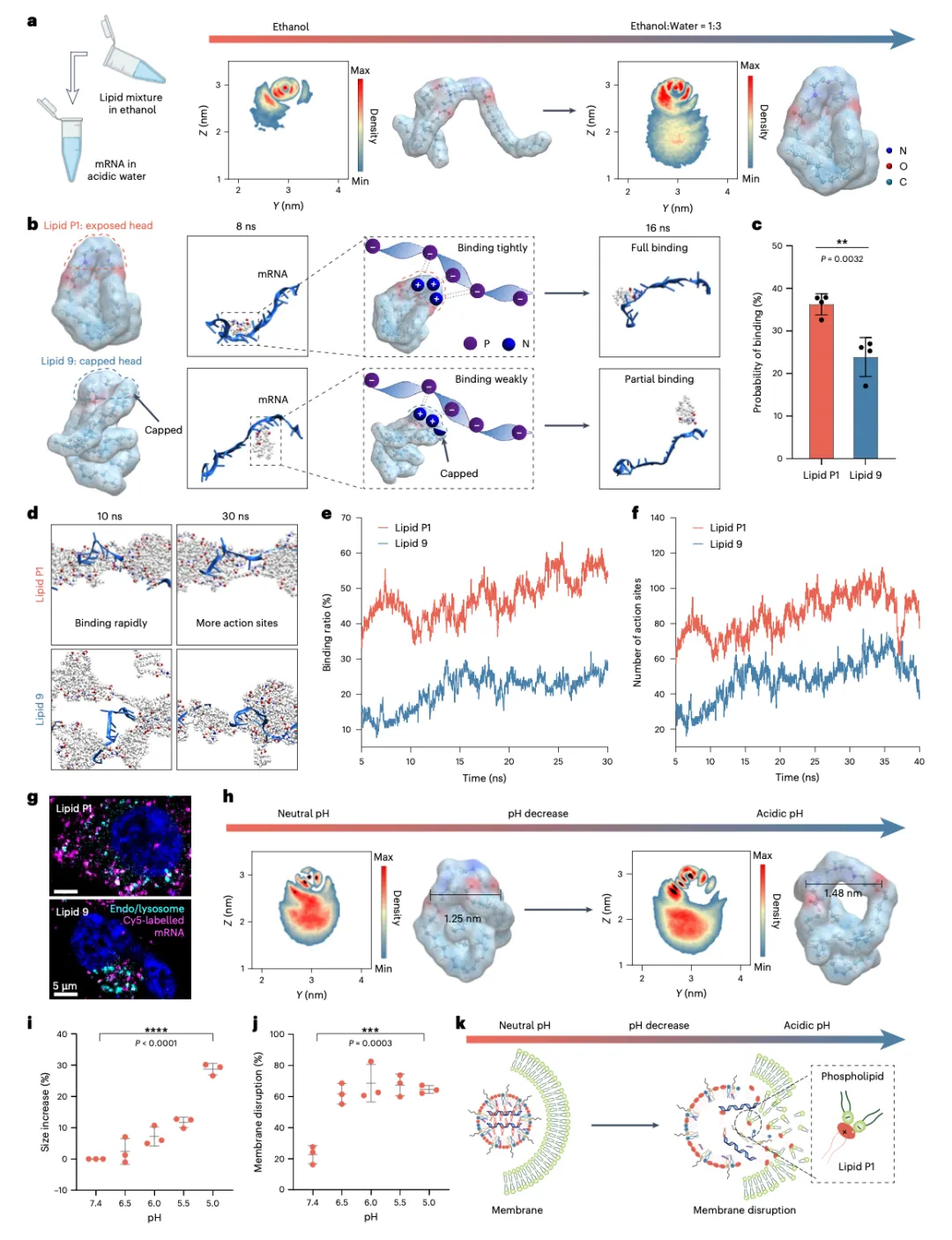
Figure 5:构象如何影响 mRNA 结合和内体逃逸
这是整篇论文的机制核心图,回答了两个关键问题:锥形构象为什么递送效率更高?构象如何影响内体逃逸?
5a 展示了 LNP 制备过程中的构象变化。 脂质 P1 在乙醇→乙醇/水混合过程中,构象变得更紧凑,头部暴露更明显。这说明 LNP 制备过程会影响脂质构象,这种变化可能影响后续的 mRNA 结合。
5b,c 对比了单个脂质与 mRNA 的结合。 Lipid P1(暴露头部)的结合概率约 38%,Lipid 9(头部被遮挡)约 25%,P = 0.0032,差异显著。结论很明确:头部暴露的脂质与 mRNA 结合概率显著更高。
5d-f 在 20 个脂质分子系统中验证了这一结论。 P1 的结合比例约 40-60%(Lipid 9 约 10-30%),结合位点约 60-100 个(Lipid 9 约 20-60 个)。锥形构象脂质确实有更多 mRNA 结合位点。
5g 用共聚焦显微镜观察内体逃逸。 品红色是 mRNA,青色是内体/溶酶体。P1 的 mRNA 与内体共定位较少(说明已逃逸到细胞质),Lipid 9 的 mRNA 与内体共定位较多(说明被困在内体中)。P1 的内体逃逸能力显著更强。
5h-j 揭示了内体逃逸的分子机制。 酸性环境下(模拟内体),脂质 P1 发生构象扩张(1.25 nm → 1.48 nm),LNP 粒径显著增加(pH 5.0 时 +28%),膜破裂能力显著增强(60-70% vs 25%)。完整机制链条是:酸性环境→脂质质子化→构象扩张→LNP 膨胀→膜破裂→mRNA 逃逸。
5k 整合了上述数据,提出完整的内体逃逸机制示意图。
Figure 5 的核心结论:锥形构象(头部暴露)→ 更强的 mRNA 结合 → 更高的递送效率;锥形构象(酸性扩张)→ 更强的膜破裂 → 更好的内体逃逸。这篇论文的价值在于,不仅证明了"构象影响功能",还解释了"为什么构象会影响功能"。
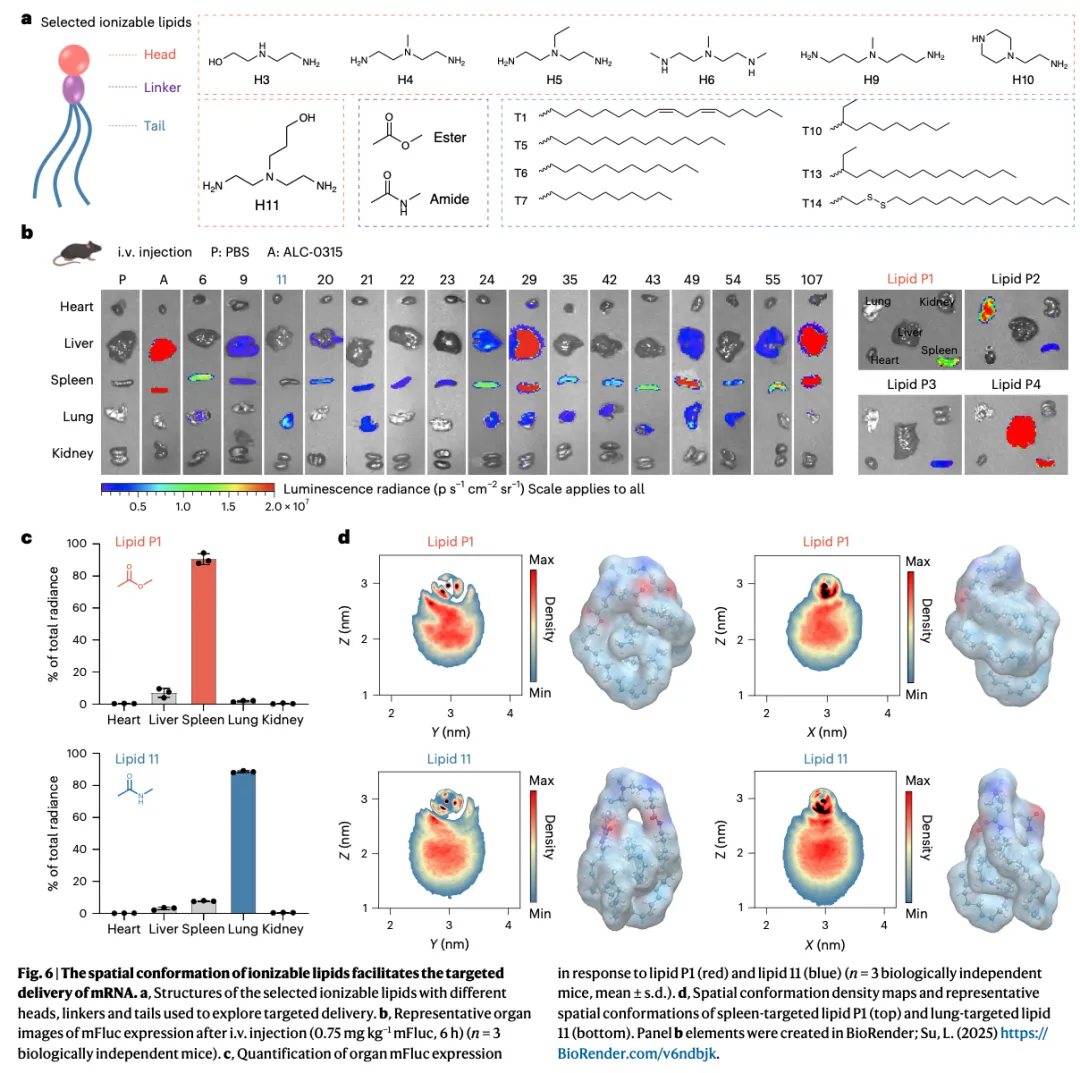
Figure 6:构象如何促进 mRNA 的靶向递送
这是器官靶向性验证图,核心结论是:不同构象的脂质会靶向不同器官。
6a 展示了筛选出的脂质结构库。 包含不同头基(H3-H10)、连接子(酯键/酰胺键)、尾部(T1-T14)的组合。这些脂质用于探索靶向递送规律。
6b 是器官分布成像图。 静脉注射后 6 小时,观察各器官的荧光素酶表达。关键发现:不同脂质靶向不同器官。P1 主要靶向脾脏(红色强信号),11 主要靶向肺部(蓝色强信号),29、35、107 等也有不同的器官分布特征。
6c 定量分析了 P1 和 11 的器官分布。 P1(酯键连接子,红色):约 90% 信号在脾脏,肝脏和肺部很少。11(酰胺键连接子,蓝色):约 90% 信号在肺部,脾脏和肝脏很少。结论:连接子类型决定器官靶向性——酯键靶向脾脏,酰胺键靶向肺部。
6d 对比了 P1 和 11 的构象密度图。 P1(脾脏靶向):构象更紧凑,头部暴露更明显。11(肺靶向):构象更伸展,头部相对被遮挡。这说明构象差异导致器官靶向性差异。
Figure 6 的核心结论:脂质的空间构象不仅影响递送效率,还决定器官靶向性。锥形构象(头部暴露,酯键)→ 脾脏靶向;伸展构象(头部遮挡,酰胺键)→ 肺靶向。这为设计器官特异性 LNP 提供了明确的指导原则。
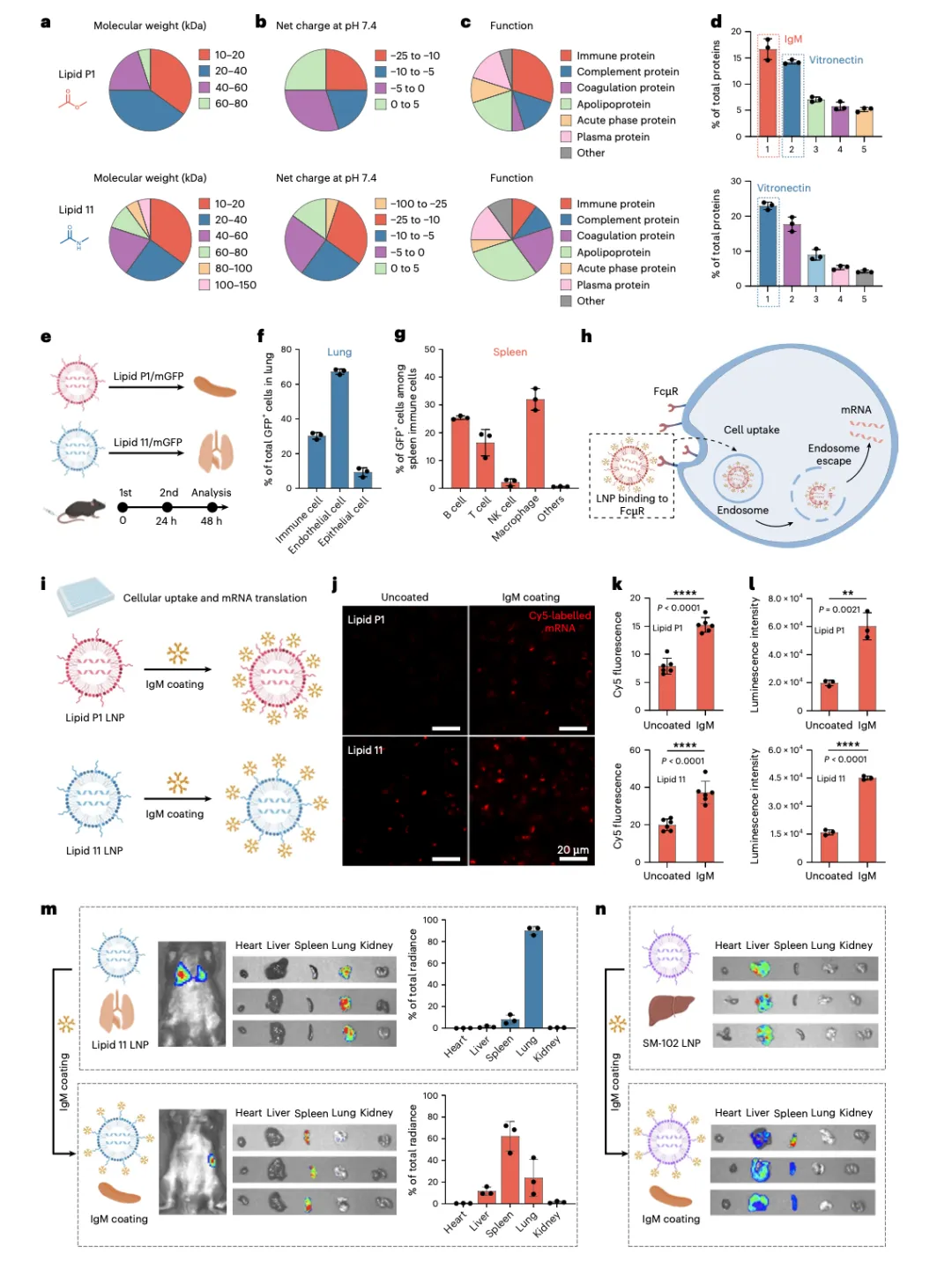
Figure 7:蛋白冠赋予 LNP 器官特异性递送能力
蛋白冠机制验证图。不同脂质吸附不同蛋白冠,蛋白冠决定器官靶向性。
7a-d 是蛋白冠蛋白质组学分析。 P1 和 11 吸附的蛋白冠组成差异显著。按功能分类:P1 吸附更多免疫蛋白(约 40%),11 吸附更多载脂蛋白(约 30%)。Top 5 蛋白中,P1 的 IgM 含量最高(约 18%),11 的 Vitronectin 最高但 IgM 很少。结论:P1 吸附 IgM,11 吸附载脂蛋白。
7e-g 用流式细胞术分析 LNP 转染的细胞类型。静脉注射 2 次后 48 小时分析。肺部:P1 和 11 主要转染免疫细胞和内皮细胞。脾脏:P1 主要转染 B 细胞(约 25%),11 转染较少。结论:P1 在脾脏优先转染 B 细胞。
7h 提出机制示意图。 IgM 包被的 LNP 通过 B 细胞表面的 FcμR 受体结合,进入细胞后内体逃逸,mRNA 翻译。这解释了 P1 为什么靶向脾脏:P1 吸附 IgM → IgM 结合 B 细胞 FcμR → LNP 被 B 细胞摄取。
7i-l 验证 IgM 包被对细胞摄取的影响。 用 Jurkat 细胞(高表达 FcμR)做实验。共聚焦成像:IgM 包被后 Cy5 荧光显著增强。定量分析:IgM 包被使 P1 的细胞摄取提高约 2 倍(P < 0.0001),mRNA 翻译提高约 3 倍(P < 0.01)。11 也有类似趋势。结论:IgM 包被通过 FcμR 显著增强细胞摄取和 mRNA 翻译。
7m 验证 IgM 包被改变器官靶向性。 Lipid 11(原本肺靶向):未包被时主要靶向肺部(约 90%),IgM 包被后转为靶向脾脏和肝脏(脾脏约 50%,肝脏约 40%)。结论:IgM 包被可以重编程器官靶向性。
7n 验证 SM-102(商业脂质,肝靶向)。 未包被时主要靶向肝脏,IgM 包被后肝脏信号降低,脾脏信号增加。结论:IgM 包被策略具有普适性。
Figure 7 的核心结论:脂质的空间构象决定吸附的蛋白冠组成,蛋白冠决定器官靶向性。P1 吸附 IgM → IgM 结合 B 细胞 FcμR → 脾脏靶向。11 吸附载脂蛋白 → 肺靶向。更重要的是,人工包被 IgM 可以重编程器官靶向性,这为设计靶向 LNP 提供了新策略。
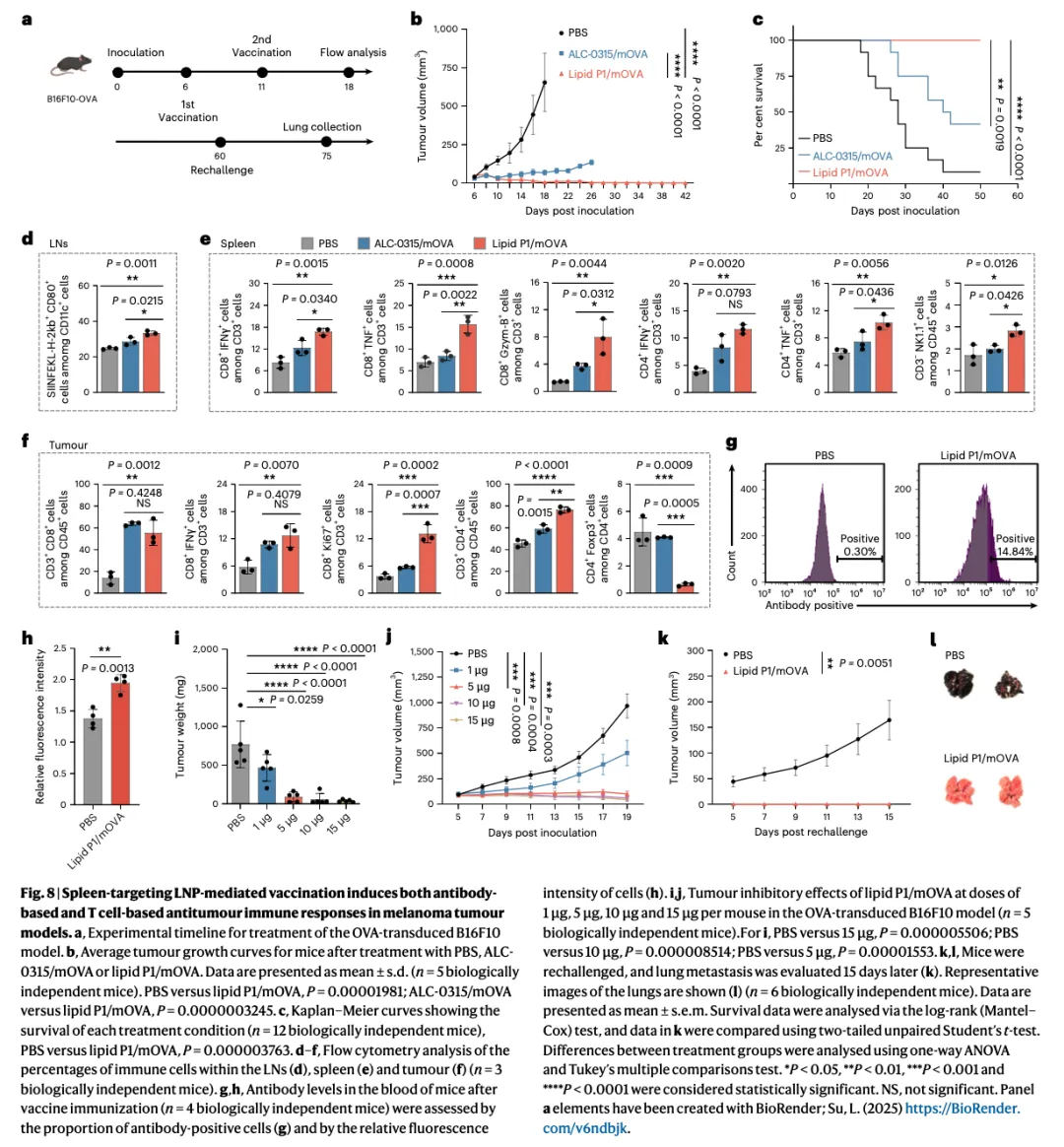
Figure 8:脾脏靶向 LNP 疫苗诱导抗肿瘤免疫反应
脾脏靶向 LNP 递送 mRNA 疫苗可以有效激活体液免疫和细胞免疫,抑制肿瘤生长。
8a 是实验时间线。 B16F10-OVA 黑色素瘤模型:第 0 天接种肿瘤,第 6 天首次疫苗注射,第 11 天加强免疫,第 18 天流式分析。另有再攻击实验:第 60 天再攻击,第 75 天收集肺部。
8b 是肿瘤生长曲线。 PBS 组肿瘤快速增长(约 750 mm³),ALC-0315 组有一定抑制(约 250 mm³),P1/mOVA 组抑制最显著(约 100 mm³)。P1 vs PBS,P < 0.0001;P1 vs ALC-0315,P < 0.0001。结论:P1 脾脏靶向疫苗显著抑制肿瘤生长。
8c 是生存曲线。 PBS 组约 30 天全部死亡,ALC-0315 组约 45 天全部死亡,P1 组约 60% 小鼠存活超过 60 天。P1 vs PBS,P < 0.0001。结论:P1 疫苗显著延长生存期。
8d-f 是流式细胞术分析免疫细胞。 淋巴结:P1 组 OVA 特异性 CD8+ T 细胞约 40%(PBS 约 25%)。脾脏:P1 组 CD8+ IFNγ+ T 细胞、CD8+ TNF+ T 细胞、CD4+ T 细胞均显著增加。肿瘤:P1 组 CD3+ CD8+ T 细胞约 70%(PBS 约 20%),Ki67+ 增殖细胞约 18%(PBS 约 5%)。结论:P1 疫苗激活了强效的 T 细胞免疫反应。
8g,h 是抗体水平检测。 PBS 组抗体阳性细胞约 0.3%,P1 组约 14.8%。相对荧光强度:P1 组约 2.0(PBS 约 1.3),P < 0.01。结论:P1 疫苗有效激活了体液免疫。
8i,j 是剂量优化实验。 1 μg、5 μg、10 μg、15 μg 四个剂量组。肿瘤重量和体积都显示:15 μg 效果最好,10 μg 次之,5 μg 和 1 μg 效果较弱。15 μg vs PBS,P < 0.0001。结论:15 μg 是最佳剂量。
8k,l 是再攻击实验和肺转移评估。 再攻击后 15 天,PBS 组肺部大量转移灶,P1 组几乎无转移。肺转移体积:P1 组约 50 mm³,PBS 组约 175 mm³,P < 0.005。结论:P1 疫苗诱导了免疫记忆,有效预防肺转移。
Figure 8 的核心结论:脾脏靶向 LNP(P1)递送 OVA mRNA 疫苗,可以有效激活体液免疫(抗体阳性细胞增加约 50 倍)和细胞免疫(CD8+ T 细胞增加约 3.5 倍),显著抑制肿瘤生长(约 85% 抑制率),延长生存期(60% 小鼠存活超过 60 天),并诱导免疫记忆预防转移。这证明了构象优化的脾脏靶向 LNP 是有效的疫苗递送平台。
Figure 5-8 的核心结论
从机制到应用的完整链条
Figure 5-8 展示了从机制理解到实际应用的完整链条:
Figure 5(mRNA 结合 + 内体逃逸机制) → Figure 6(器官靶向验证) → Figure 7(蛋白冠介导机制) → Figure 8(疫苗应用验证)
构象 → mRNA 结合/内体逃逸 → 器官分布 → 蛋白冠介导 → 免疫反应 → 肿瘤抑制
这个完整的链条证明了:
构象优化不仅可以提高递送效率,还可以实现器官特异性靶向,进而激活特定的免疫反应,最终实现治疗效果。
八、这篇文章真正的科学贡献不是 AI
如果只从机器学习角度看,这篇会被低估。
它最重要的部分,其实是下面这些规律:
构象与 mRNA 结合
分子动力学模拟显示:锥形(头部暴露)与 mRNA 结合更稳定,头部被尾部遮挡则结合不稳定。
构象与内体逃逸
在酸性环境下:头部质子化导致扩张,促进膜插入与破坏。锥形结构更容易产生这种变化。
构象与器官靶向
不同脂质改变蛋白冠组成,进而改变器官分布。
例如:P1(酯键连接子)靶向脾脏,11(酰胺键连接子)靶向肺部。
不是"结构直接决定器官",而是一个中介过程:
构象 → 蛋白冠 → 器官分布
有没有机制解释?还是只是相关性?
构象影响蛋白冠(蛋白质组学验证),蛋白冠介导器官靶向(IgM 富集到脾脏),机制链条完整。
九、如果你是审稿人,会真正卡它什么?
这不是"构象建模",而是"构象投影"
作者做的是:
2000 多个构象 → 对齐 → 3D 密度图 → 2D 密度图 → 28 个特征 → 5 个关键特征
问题不是"对不对",而是这个投影是否保留了决定功能的那部分信息?
如果关键差异存在于被压缩掉的维度中,模型将永远学不到。
单分子构象,是否能代表 LNP 行为?
这篇的隐含假设是单个脂质的构象决定 LNP 性能。
但实际系统是多分子自组装、动态重排、蛋白冠参与。
单分子构象,是原因,还是代理变量(proxy)?
更进一步的问题是:
模型学到的是"构象本身",还是"构象与标签之间的实验相关性"?
如果蛋白冠、细胞类型、实验条件没有严格控制:
模型可能只是学到:某类构象在某个实验体系下更有效,而不是普适规律。
标签是否"可学习"?
模型预测的是递送效率。
但这个标签本身来源于异质性群体、实验噪声高、批次差异明显。
当标签是分布时,误差下限是"物理决定的",不是模型决定的。
十、一个更值得思考的问题
这篇文章已经往前走了一步:从结构走向构象。
但仍然有一个没有完全解决的问题:
LNP 本身是一个分布,而不是一个确定结构。
单分子:构象分布(这篇考虑了)。
单颗粒:结构异质性(这篇未考虑)。
群体:群体分布(这篇未考虑)。
而当前机器学习仍在学习一个"平均表示"。
这可能解释一个现象:
为什么很多 LNP 的机器学习模型,R 平方很难超过0.9。
因为训练数据的"标签"(如递送效率)本身是一个分布,但模型学习的是"平均值"。
分布的方差,就是模型的误差下限。
这是未来 LNP 与 AI 交叉领域需要突破的方向:
从"单点预测"走向"分布预测",从"平均表示"走向"异质性建模"。
结尾
如果从 AI 的角度看,这篇文章是克制的:
没有使用复杂模型,没有追求极致指标,也没有过度宣传预测能力。
但它做了一件更重要的事情:
把一个长期被忽略的变量——构象——引入到了可计算框架中。
更值得注意的是,它也暴露了一个更深层的问题:
当前 LNP 与 AI 交叉领域的瓶颈,可能不在模型,而在表示。
我们仍然在用单一结构、平均特征、单点标签,去描述一个本质上是分布、异质性、动态系统。
如果这个前提不改变,模型再复杂,提升也会越来越有限。
这篇文章真正留下一个问题:
下一步,LNP 与 AI 应该如何从"平均表示"走向"分布建模"?
当我们开始讨论"分布",而不是"单一结构"时,LNP 与 AI 交叉领域才刚刚开始。
参考文献
Lin-Jia Su, Nan-Nan Wang, Rui Luo, et al. Artificial intelligence-guided design of LNPs for in vivo targeted mRNA delivery via analysis of the spatial conformation of ionizable lipids. Nature Biomedical Engineering. 2026. doi:10.1038/s41551-026-01640-8
Take home message
先看变量,而不是模型
模型只能优化你定义的问题。
先看表示,而不是算法
模型只学习你给它的信息。
先看外推,而不是精度
不能预测未知的数据,模型没有意义。
LNP 与 AI/ML 系列推文
LNP x AI/ML|为什么 LNP 终于开始需要新的方法,而经验优化正在接近边界
LNP x AI/ML|同样做 LNP,为什么有些实验数据能建模,有些永远只能停留在经验里?
LNP x AI/ML|为什么很多 AI 推荐的 LNP 配方,回到实验室就失效?
LNP x AI/ML|为什么 SHAP 图里最重要的变量,不一定是真正决定 LNP 递送的变量
智能递送
