 夜雨聆风
夜雨聆风下载所有乳酸菌基因组并且做基因预测-20260415
1,背景介绍
2020年(Zheng et al. 2020)乳杆菌科(Lactobacillaceae)分类大调整的核心,是基于全基因组序列分析(核心基因组系统发育、平均核苷酸/氨基酸一致性、特征基因、生态与代谢特性等),将原先高度异质的Lactobacillus属(当时约261种)拆分成25个属(包括保留并修正描述的Lactobacillus属、Paralactobacillus属,以及23个新属)。同时,将原Lactobacillaceae和Leuconostocaceae两科合并为一个科(现在的Lactobacillaceae),整体属的数量一度达到约31个(含Pediococcus、Weissella、Leuconostoc、Oenococcus、Fructobacillus、Convivina等原属)。
保留/修正的属:
Lactobacillus(乳酸菌属,模式种:L. delbrueckii)——主要保留宿主适应型物种(如L. acidophilus、L. crispatus、L. gasseri、L. johnsonii、L. iners等,适应脊椎动物或昆虫肠道/阴道等)。 Paralactobacillus(副乳杆菌属)——少数物种。
23个新属(多数名称以“L”开头,便于过渡记忆,原种名通常保留):
Acetilactobacillus(醋酸乳杆菌属) Agrilactobacillus(农乳杆菌属) Amylolactobacillus(淀粉乳杆菌属) Apilactobacillus(蜜蜂乳杆菌属,常与昆虫相关) Bombilactobacillus(蜂乳杆菌属,与蜂蜜/蜜蜂相关) Companilactobacillus(伴生乳杆菌属) Dellaglioa(德拉吉奥乳杆菌属) Fructilactobacillus(果糖乳杆菌属) Furfurilactobacillus(糠乳杆菌属) Holzapfelia(霍尔扎普费尔乳杆菌属) Lacticaseibacillus(干酪乳杆菌属,如原L. casei、L. paracasei、L. rhamnosus等,常见于发酵乳制品和益生菌) Lactiplantibacillus(植物乳植杆菌属,即你之前提到的“植物乳杆菌”,模式种:L. plantarum,适应植物/发酵食品) Lapidilactobacillus(石乳杆菌属) Latilactobacillus(宽乳杆菌属,如L. sakei等) Lentilactobacillus(透镜乳杆菌属,如原L. buchneri群) Levilactobacillus(短乳杆菌属,如L. brevis等,异型发酵) Ligilactobacillus(结肠乳杆菌属/唾液乳杆菌群部分) Limosilactobacillus(粘液乳杆菌属,即你说的“limo乳杆菌”,如L. fermentum、L. reuteri等,许多能产胞外多糖,常见益生菌) Liquorilactobacillus(酒乳杆菌属/唾液乳杆菌群部分) Loigolactobacillus(洛伊戈乳杆菌属,如原L. coryniformis群) Paucilactobacillus(少乳杆菌属) Schleiferilactobacillus(施莱费尔乳杆菌属) Secundilactobacillus(次乳杆菌属)
所以,现在的乳酸菌属,只有Lactobacillus了。不过我关注的包括植物乳杆菌,就一切下载:
乳酸菌属:Lactobacillus
植物乳杆菌属:Lactiplantibacillus
2,下载基因组
先下载4种常见的乳酸菌的参考基因组。
安装下载软件:
mamba install ncbi-datasets-cli -y这是NCBI官方的软件。
获取Lactobacillus属和Lactiplantibacillus属下面的种名字:
安装软件:
#运行的命令里用了 jq(一个 JSON 处理工具)mamba install jq需要用到dataformat,不过通常和 datasets 一起装好的,不用额外安装。
逻辑:现在NCBI里看,属于这两个属的菌种有哪些,然后按照细菌的种名下载。
代码:
#!/bin/bash# =============================================# 提取并清洗 Lactobacillus 和 Lactiplantibacillus 的物种列表# 只保留 "Genus species" 格式,去掉 strain、DSM、ATCC 等后缀# =============================================set -eecho"=== Step 1: 提取原始物种列表 ==="echo"正在获取 Lactobacillus 所有物种..."datasets summary genome taxon "Lactobacillus" --as-json-lines > Lactobacillus_species.jsonlcat Lactobacillus_species.jsonl | dataformat tsv genome --fields organism-name | tail -n +2 | sort | uniq > Lactobacillus_raw.txtecho"正在获取 Lactiplantibacillus 所有物种..."datasets summary genome taxon "Lactiplantibacillus" --as-json-lines > Lactiplantibacillus_species.jsonlcat Lactiplantibacillus_species.jsonl | dataformat tsv genome --fields organism-name | tail -n +2 | sort | uniq > Lactiplantibacillus_raw.txtecho"=== Lactobacillus_raw.txt 前 5 行示例 ==="head -n 5 Lactobacillus_raw.txt | cat -necho"=== Lactiplantibacillus_raw.txt 前 5 行示例 ==="head -n 5 Lactiplantibacillus_raw.txt | cat -n# ====================== 清洗函数:只保留 Genus species ======================clean_species_list() {local input_file="$1"local output_file="$2"local genus="$3"# Lactobacillus 或 Lactiplantibacillusecho"正在清洗 $input_file → $output_file ..." cat "$input_file" | \# 1. 去掉 Candidatus(不管大小写) grep -v -i "^Candidatus" | \# 2. 去掉未定种 (sp.) grep -v -i " sp\." | \# 3. 去掉 unclassified 或 metagenome 等明显杂项 grep -v -i "unclassified\|metagenome\|uncultured" | \# 4. 只保留包含属名的行(宽松匹配,防止把正常行也干掉) grep -i "$genus" | \# 5. 取前两个字段(Genus species),忽略后面的 strain/subsp./DSM 等 awk '{print $1, $2}' | \# 6. 去重 + 只保留正好两个词的行 sort | uniq | \ awk 'NF == 2 && $1 ~ /^[A-Z]/' > "$output_file"}clean_species_list "Lactobacillus_raw.txt""Lactobacillus_species_list.txt""Lactobacillus"clean_species_list "Lactiplantibacillus_raw.txt""Lactiplantibacillus_species_list.txt""Lactiplantibacillus"# 显示结果echo"=== Lactobacillus 干净物种数量 ==="wc -l Lactobacillus_species_list.txtecho"前 5 个示例:"head -n 5 Lactobacillus_species_list.txtecho"=== Lactiplantibacillus 干净物种数量 ==="wc -l Lactiplantibacillus_species_list.txtecho"前 5 个示例:"head -n 5 Lactiplantibacillus_species_list.txtecho"✅ 清洗完成!干净列表已保存为 *_species_list.txt"echo" (只保留属名 + 种名,例如:Lactiplantibacillus pentosus)"结果:
(bio) zhoukc@dell-PowerEdge-R750xs:~/Bac_genome/test2$ bash 1.sh === Step 1: 提取原始物种列表 ===正在获取 Lactobacillus 所有物种...正在获取 Lactiplantibacillus 所有物种...=== Lactobacillus_raw.txt 前 5 行示例 === 1 Candidatus Lactobacillus pullistercoris 2 Candidatus Paralactobacillus gallistercoris 3 Lactobacillus acetotolerans 4 Lactobacillus acetotolerans DSM 20749 = JCM 3825 5 Lactobacillus acidophilus=== Lactiplantibacillus_raw.txt 前 5 行示例 === 1 Lactiplantibacillus argentoratensis 2 Lactiplantibacillus argentoratensis DSM 16365 3 Lactiplantibacillus brownii 4 Lactiplantibacillus carotarum 5 Lactiplantibacillus daoliensis正在清洗 Lactobacillus_raw.txt → Lactobacillus_species_list.txt ...正在清洗 Lactiplantibacillus_raw.txt → Lactiplantibacillus_species_list.txt ...=== Lactobacillus 干净物种数量 ===51 Lactobacillus_species_list.txt前 5 个示例:Lactobacillus acetotoleransLactobacillus acidophilusLactobacillus agrestimurisLactobacillus amylolyticusLactobacillus amylovorus=== Lactiplantibacillus 干净物种数量 ===20 Lactiplantibacillus_species_list.txt前 5 个示例:Lactiplantibacillus argentoratensisLactiplantibacillus browniiLactiplantibacillus carotarumLactiplantibacillus daoliensisLactiplantibacillus daowaiensis✅ 清洗完成!干净列表已保存为 *_species_list.txt (只保留属名 + 种名,例如:Lactiplantibacillus pentosus)下载Lactobacillus属和Lactiplantibacillus属下面所有种的的参考基因组:
一共71个细菌的基因组。
#!/bin/bash# ================== 从清洗后的列表文件动态读取物种 ==================echo"=== 从列表文件加载物种 ==="# 合并两个干净列表(去重 + 排序)cat Lactobacillus_species_list.txt Lactiplantibacillus_species_list.txt 2>/dev/null | \ sort | uniq > all_species_list.txt# 构建 species 数组species=()while IFS= read -r line || [[ -n $line ]]; doif [[ -n "$line" && "$line" =~ ^[A-Z] ]]; then species+=("$line")fidone < all_species_list.txtecho"共加载 ${#species[@]} 个物种,将下载它们的参考基因组..."# 如果列表为空,给个友好提示if [ ${#species[@]} -eq 0 ]; thenecho"警告:列表为空!请检查 Lactobacillus_species_list.txt 和 Lactiplantibacillus_species_list.txt 是否有内容。"exit 1fifor sp in"${species[@]}"; doecho"正在处理 ${sp}..." sp_name="${sp// /_}" output_dir="${sp_name}_genome" mkdir -p "$output_dir"# 下载参考基因组(只下载 reference + genome 文件) datasets download genome taxon "${sp}" \ --reference \ --filename "${sp_name}.zip" \ --include genomeif [ ! -f "${sp_name}.zip" ]; thenecho"错误: ${sp} 下载失败(可能没有 reference 基因组)"continuefi# 解压 unzip -q "${sp_name}.zip" -d "${sp_name}_tmp"# 重命名文件(保留 accession) find "${sp_name}_tmp/ncbi_dataset/data" -name "*.fna" | whileread -r file; do accession=$(basename $(dirname "$file")) cp "$file""$output_dir/${sp_name}_${accession}.fa"done# 清理 rm -rf "${sp_name}.zip""${sp_name}_tmp"echo"${sp} 完成 ($(ls "$output_dir"/*.fa 2>/dev/null | wc -l) 个基因组)"doneecho"所有物种处理完成!"结果:
(bio) zhoukc@dell-PowerEdge-R750xs:~/Bac_genome/test2$ bash 2.sh === 从列表文件加载物种 ===共加载 71 个物种,将下载它们的参考基因组...正在处理 Lactiplantibacillus argentoratensis...Collecting 1 genome record [================================================] 100% 1/1Downloading: Lactiplantibacillus_argentoratensis.zip 904kB valid data packageValidating package files [================================================] 100% 5/5Lactiplantibacillus argentoratensis 完成 (1 个基因组)正在处理 Lactiplantibacillus brownii...Collecting 1 genome record [================================================] 100% 1/1Downloading: Lactiplantibacillus_brownii.zip 931kB valid data package......#结束后计数一下(bio) zhoukc@dell-PowerEdge-R750xs:~/Bac_genome/test2$ ls */*fa | wc -l69下载了69个,应该有2个基因组没有参考基因组或者下载失败,无所谓了。
3,基因预测-prodigal
基因预测:
安装软件:
mamba install prodigalprodigal的翻译:败家子,哈哈,这名字叫得
关键参数详解
-p meta应用场景:处理宏基因组数据(短序列、混合样本)。 与 single区别:meta模式不依赖物种特异性信号(如SD序列),适合多样性高的样本。-g(密码子表)11:标准细菌密码子(默认)。4:支原体/低GC含量菌(如TGA编码色氨酸而非终止子)。常见选项: 重要性:错误选择会导致基因预测不准确(如起始/终止位点错误)。 -f gff输出内容:基因坐标、链方向、功能注释(如 ID=1_1;partial=00表示完整基因)。下游分析:可直接用于基因组浏览器(如IGV)或注释工具(如Prokka)。 -t(训练文件)适用情况:默认模型在极端GC含量或特殊密码子使用物种表现不佳时,需自定义训练。
4,蛋白注释-eggnog_mapper
安装数据库:
#python 小于3.7,eggnog-mapper不支持mamba create -n eggnog_mapper python=3.10 -y eggnog-mapper#验证mamba run -n eggnog_mapper emapper.py --helpmamba install -y eggnog-mapper# 创建数据库目录(确保有写入权限)mkdir -p /home/zhoukc/eggnog_db# 下载数据库(指定具体路径),数据有点大,建议挂后台下载nohup download_eggnog_data.py -y --data_dir /home/zhoukc/eggnog_db &(bio) zhoukc@dell-PowerEdge-R750xs:~/eggnog_db$ du -h *39G eggnog.db8.7G eggnog_proteins.dmnd266M eggnog.taxa.db6.4M eggnog.taxa.db.traverse.pkl18M nohup.out差不多50G左右。
把69个基因组,放在一个文件夹中:
mkdir Lactobacillus_Lactiplantibacillus_69mv *genome/*fa ./Lactobacillus_Lactiplantibacillus_69ls Lactobacillus_Lactiplantibacillus_69 | wc -lrm -rf *genomelscd Lactobacillus_Lactiplantibacillus_69功能注释:
#!/bin/bash# 全局设置THREADS=64 # CPU线程数EGGNOG_DATA="/home/zhoukc/eggnog_db"# eggNOG数据库路径LOG_FILE="annotation_batch.log"# 日志文件# 清空旧日志> "$LOG_FILE"# 防止没有匹配文件时把 *.fa 当成字面文件名shopt -s nullglob# 循环处理所有 .fa 基因组文件for GENOME_FILE in *.fa; do# 提取前缀(去掉 .fa 后缀) SAMPLE_PREFIX="${GENOME_FILE%.fa}"echo"Processing $GENOME_FILE | Sample: $SAMPLE_PREFIX" | tee -a "$LOG_FILE"echo"----------------------------------------" | tee -a "$LOG_FILE"# ===== 1. 基因预测 (Prodigal) ===== GFF_OUT="${SAMPLE_PREFIX}_genes.gff" PROTEIN_OUT="${SAMPLE_PREFIX}_proteins.faa" SCORES_OUT="${SAMPLE_PREFIX}_gene_scores.txt"echo"[1/2] Running Prodigal..." | tee -a "$LOG_FILE" prodigal \ -i "$GENOME_FILE" \ -o "$GFF_OUT" \ -a "$PROTEIN_OUT" \ -f gff \ -g 11 \ -p single \ -q \ -s "$SCORES_OUT" \ >> "$LOG_FILE" 2>&1if [ ! -s "$PROTEIN_OUT" ]; thenecho"ERROR: Prodigal failed or produced empty $PROTEIN_OUT" | tee -a "$LOG_FILE"exit 1fi# ===== 2. 功能注释 (eggNOG-mapper) ===== EGGNOG_OUT="${SAMPLE_PREFIX}_eggNOG"echo"[2/2] Running eggNOG-mapper..." | tee -a "$LOG_FILE" mamba run -n eggnog_mapper emapper.py \ -i "$PROTEIN_OUT" \ -o "$EGGNOG_OUT" \ --cpu "$THREADS" \ --sensmode fast \ --data_dir "$EGGNOG_DATA" \ --tax_scope auto \ --report_orthologs \ >> "$LOG_FILE" 2>&1echo"Annotation completed for $SAMPLE_PREFIX" | tee -a "$LOG_FILE"echo"----------------------------------------" | tee -a "$LOG_FILE"doneecho"ALL SAMPLES PROCESSED. Check $LOG_FILE for details." | tee -a "$LOG_FILE"过程:
(bio) zhoukc@dell-PowerEdge-R750xs:~/Bac_genome/test2/Lactobacillus_Lactiplantibacillus_69$ tail -f nohup.out Processing Lactiplantibacillus_argentoratensis_GCF_037290055.1.fa | Sample: Lactiplantibacillus_argentoratensis_GCF_037290055.1----------------------------------------[1/2] Running Prodigal...[2/2] Running eggNOG-mapper...Annotation completed for Lactiplantibacillus_argentoratensis_GCF_037290055.1----------------------------------------Processing Lactiplantibacillus_brownii_GCF_045862825.1.fa | Sample: Lactiplantibacillus_brownii_GCF_045862825.1----------------------------------------[1/2] Running Prodigal...[2/2] Running eggNOG-mapper...一个基因组差不多3min吧。
每个基因组7个文件。
Prodigal 生成了: .proteins.faa、.genes.gff、.gene_scores.txteggNOG-mapper 生成了: .emapper.annotations、.emapper.hits、.emapper.orthologs、.emapper.seed_orthologs
1. Prodigal 生成的文件(基因预测阶段)
Lactiplantibacillus_xxx_proteins.faa用途:最重要的文件之一 这是从基因组DNA中预测出来的所有蛋白质序列(氨基酸序列,FASTA格式)。 eggNOG-mapper 就是拿这个文件去做功能注释的。 后续分析最常用:你做代谢通路、毒力因子、核心基因组分析、蛋白结构预测等,几乎都要用到这个 .faa 文件。 Lactiplantibacillus_xxx_genes.gff用途:基因位置注释文件 用 IGV、Artemis 等软件可视化看基因位置 做基因组比较(Pan-genome)时需要 提取特定区域的序列 记录了每个预测基因在基因组上的精确位置(起始位点、终止位点、链的方向、正负链等)。 格式是标准的 GFF3。 常用场景: Lactiplantibacillus_xxx_gene_scores.txt用途:Prodigal 的内部打分文件 记录了每个预测基因的置信度分数(score)。 一般不常用,主要用于调试或你想严格筛选“高置信基因”的时候。 大部分情况下可以直接删除节省空间。
2. eggNOG-mapper 生成的文件(功能注释阶段)
Lactiplantibacillus_xxx_eggNOG.emapper.annotations用途:最重要、最核心的结果文件(强烈建议保留) 每个蛋白的 eggNOG ortholog(OG) 功能描述(Description) GO terms(基因本体,分子功能、生物过程、细胞组分) KEGG 通路(ko 号) COG 分类 EC number(酶 commission 号) PFAM 结构域等 这是你跑 eggNOG-mapper 后最主要的输出。 内容包括: 后续分析基本都依赖这个文件:做功能富集、代谢网络构建、比较不同菌株的功能差异等。 Lactiplantibacillus_xxx_eggNOG.emapper.hits用途:比对的原始 hits 文件 记录了每个蛋白在 eggNOG 数据库中最相似的种子序列(seed ortholog)的比对信息(类似 blast 结果)。 包含 score、e-value、identity 等。 一般不直接使用,主要用于调试或你想查看具体比对细节时。 Lactiplantibacillus_xxx_eggNOG.emapper.seed_orthologs用途:种子直系同源物列表 列出了每个蛋白匹配到的 eggNOG “seed ortholog” 的 ID。 中等重要,有时候你想进一步用 eggNOG online 工具深入分析时会用到。 Lactiplantibacillus_xxx_eggNOG.emapper.orthologs用途:更完整的直系同源群信息 记录了该蛋白所属的 orthologous group(OG)包含的其他物种成员。 可选保留,做进化分析或比较基因组学时比较有用,普通功能注释可以不保留。
实际使用建议(针对你的 69 个 Lactobacillus/Lactiplantibacillus 菌株)
强烈推荐保留的文件(每个菌株留这 2 个就够了):
*_proteins.faa(蛋白序列)*_eggNOG.emapper.annotations(功能注释表)
可以安全删除的文件(节省磁盘空间):
*_gene_scores.txt*_genes.gff(除非你需要看基因位置或做可视化)*_eggNOG.emapper.hits*_eggNOG.emapper.seed_orthologs*_eggNOG.emapper.orthologs
5,查找自己关注的酶
一个细菌(尤其是乳酸菌,如Lactobacillus、Lactococcus 等)能分泌(或产生并释放)乳酸,主要依赖乳酸发酵(lactic acid fermentation)途径。乳酸不是细菌主动“分泌”的次级代谢物,而是糖酵解(glycolysis)途径的终产物,通过细胞膜上的转运蛋白(如乳酸/质子共转运蛋白)排出到胞外,导致培养基酸化。
核心酶:乳酸脱氢酶(Lactate Dehydrogenase, LDH) 酶的编号:EC 1.1.1.27或者EC 1.1.1.28
EC 号:
EC 1.1.1.27:L-乳酸脱氢酶(L-lactate dehydrogenase),催化产生 L-乳酸(大多数乳酸菌产生此异构体)。 EC 1.1.1.28:D-乳酸脱氢酶(D-lactate dehydrogenase),催化产生 D-乳酸(部分乳酸菌产生此异构体,或同时产生两种)
需要找的酶:
简单查看:
grep "1.1.1.27" -w *eggNOG.emapper.annotations | awk -F '\t''{print $1,$3,$4, $9, $11}' | column -t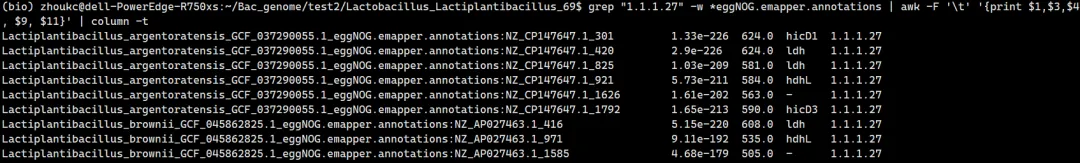
第一列是query_name
第三列是evalue,通常认为 evalue < 1e-3 是可信匹配,< 1e-10 为高度可信。
第四列是score,一般 >50 即可信
第九列是Preferred_name
第11列是EC编号
evalue | score | |
|---|---|---|
| 关注点 | ||
| 依赖性 | ||
| 应用场景 | ||
| 用户选择 | evalue | score |
evalue:判断匹配是否随机,越小越好(重点关注)。score:衡量匹配质量,越大越好(跨数据库比较时有用)
接下来就是自己按照需求可视化了,R语言里做个统计柱状图,基因有无得热图啥的,那就手拿把掐了。
