 夜雨聆风
夜雨聆风一句话总结:2026年4月,Team-NB发布MDR技术文档提交最佳实践指南V4版。92页内容来自20+家公告机构的真实审核经验——他们把审评中最常见的不符合项全部摆在了台面上。这是国内制造商不可错过的一手信息。

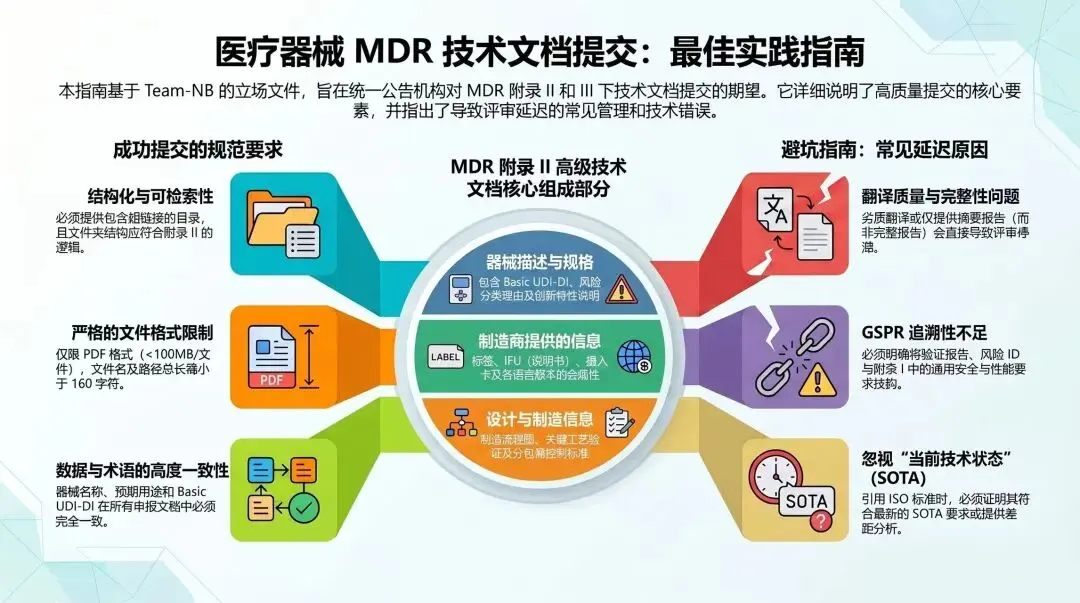
2026年4月21日,Team-NB(欧洲公告机构协会)发布了《MDR技术文档提交最佳实践指南》V4版(Best Practice Guidance, 以下简称“BPG”)。92页的厚度,由20+家公告机构联合编写,覆盖Annex II和Annex III的全部要素。
这不是一份法规,也不是协调标准。但如果你想知道公告机构的审核员坐在会议室里,翻开你的技术文档时,第一眼会扫哪里、最在意什么、最常在哪些地方扣分——这份指南就是唯一的参考答案。每一条都来自真实审核经验,每一条都是中国制造商的"扣分重灾区"。
一、14条致命缺陷
缺陷 1:Intended Purpose在各个文档中"各自为政"
Team-NB立场:"同一款器械的Intended Purpose在CEP、CER、DoC、IFU、标签中出现不同表述,是我们最常在第一次审评时发现的Bug。"
这是最基础、也最容易犯的错误。审核员会交叉检查所有文档中的Intended Purpose是否一致。不一致 → 观察项。
解决:建立"关键定义锁定机制"——把Intended Purpose、Indications、Contraindications、Target Population写在一个"金标准"文件中,所有文档引用同一个版本。
缺陷 2:GSPR合规矩阵写"NA"却不解释
Team-NB立场:"GSPR矩阵里写不适用(NA)是最容易的,但也是最不能接受的。你告诉我为什么这个条款不适用于你的器械。"
MDR Annex I 有23条GSPR,其中很多条款对一些器械确实不适用。但仅仅在表格里写NA是不够的——你必须给出理由。
正确格式:使用5列表格——GSPR条款、是否适用、理由、采用的方法/标准、引用文件。
缺陷 3:风险管理只说"写进IFU"就完事
Team-NB立场:"如果一个风险的控制措施仅仅是'在IFU中警告',而没有证据证明这个警告对使用者有效,我们会认为这个风险没有被充分控制。"
MDR Annex II 5(b)明确要求:风险控制措施必须验证有效性。如果你说"用户操作前须阅读IFU",那么在可用性工程报告(EN 62366-1)中必须有证据证明这个警告在预期用户群体中是有效的。
缺陷 4:同一份信息在多处出现却互相矛盾
Team-NB立场:"同一个验证报告的编号在TD的两个地方不一样,被发现了就是不符合项。我们理解这种疏忽,但我们不能放过——因为这意味着制造商的文档管理系统有问题。"
审核员不是一个人在审文档。一个看风险部分,另一个看验证部分。两份报告引用了不同的版本号——他们回到评审圆桌上一对——发现不一致。
基本规矩:所有引用关系保持单一来源。用一个版本号管理机制,确保所有文件引用的报告是最新版本。
缺陷 5:灭菌验证——教科书级的错误
对中国制造商尤其需要注意的点:
环氧乙烷(ETO)灭菌验证报告中,未考虑儿科使用场景的ETO残留限值 重力下排气式蒸汽灭菌方案——被BPG明确指出不视为SOTA(最先进技术) 验证文件中缺失产品族识别理由(为什么用这个最差情况产品来代表整个产品族) 常规生物负载测试频率不足或方法不当
缺陷 6:可复用器械(Class Ir)——再处理验证的"盲区"
📌 和中国出口制造商最直接相关
BPG明确要求:IFU中的每一个再处理声明(清洁、消毒、包装、灭菌)都须有验证;功能测试必须覆盖最大复用次数;过程残留物累积(清洁剂、消毒剂、ETO等)须纳入生命周期验证;重力置换蒸汽灭菌不被视为SOTA。
换句大白话:如果你的器械可以复用50次,你的验证数据必须证明第50次使用时,产品性能和生物相容性依然合格。
缺陷 7:生物相容性评估——程序合规≠结果被接受
最常见的问题:
生物评估人员/毒理学家的资质证据缺失(不只是一张简历,而是能证明该人员有相关器械评估经验的文档) 测试实验室的ISO/IEC 17025认证在测试时点已过期 - 接触时间累积计算错误
——MDR要求考虑累计暴露时间,不是单次接触
ISO 10993-1:2025已是最新版本,请确认你引用的不是旧版。
缺陷 8:软件V&V——测试数据不够,还说"够"
对含软件的器械,NB重点审:
训练集和测试集是否独立(AI/ML器械尤其) 异常值处理——所有异常值须有根因分析 已知残留异常清单——不得隐藏,须逐一说明可接受理由
缺陷 9:网络安全——不再是"锦上添花"
从BPG V4开始,网络安全文档的细节要求显著增加:
须提供安全开发生命周期证据(计划、评估、控制措施验证) 威胁建模方法须明确(STRIDE、攻击面分析等) SOUP漏洞的持续监控管理 覆盖机密性、完整性、可用性三原则(MDCG 2019-16)
缺陷 10:PSUR只是一个"数据汇编"而非"分析"
Team-NB立场:"很多PSUR把投诉、警戒、维护报告全部罗列了一遍,但从来没有得出结论——这个器械的受益-风险比有没有变化?"
PSUR的使命不是展示数据,而是基于数据做分析。
要用比较分析:今年的数据相比去年趋势如何? 要下结论:受益-风险比是否持续可接受? 要反馈SOP:结论是否影响了SSCP?风险管理文件?临床试验计划?
缺陷 11:PMS计划没有区分反应性和主动性
PMS计划中,NB会区分两类数据来源进行评估:
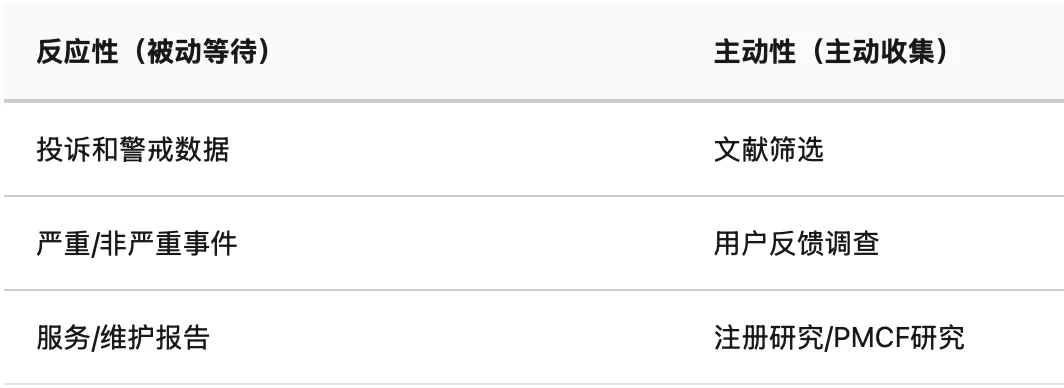
很多制造商的PMS计划只有反应性活动。BPG非常直白地指出——所有器械都需要主动性一般PMCF。
缺陷 12:再处理验证——水的质量没说清楚
Team-NB立场:"你说最终漂洗用水是'去离子水'——这远远不够。它的微生物指标是什么?化学纯度标准是什么?"
最终漂洗水的微生物质量和化学纯度须按照国际标准或药典来定义。只说"去离子水"或"纯化水"在没有明确引用标准的情况下,审核员会给出不符合项。
缺陷 13:文件格式不规范——还没开始就输了
你可能觉得"形式不重要"——但NB在BPG中专门强调:
单个PDF文件不超过100MB 文件路径(含文件名)不超过160个字符 - 必须设置书签(Bookmarks),让审核员能在长文档中快速导航
PDF须是可搜索的(OCR,如果是扫描件) 文件名中应包含文档类型和版本号
⚠️ 重要:这条不是法规要求,但BPG说得很直白:不规范的文件格式会直接导致审评延迟。一个无法搜索、没有书签的200MB PDF——审核员可能直接退回。
缺陷 14:临床评价——Equivalent Device的证据链断裂
对中国制造商来说,Equivalent声明是最常见的临床评价路径。但这条路径有三个严格的证据前提:
制造商须拥有访问等效设备技术文档的合同协议(MDCG 2023-7) Class III/植入式器械——Equivalent声明必须有充分的科学证据支持 若使用了Article 61(10)例外路径,却同时又计划了全面的PMCF临床研究——矛盾声明(因为61(10)的前提是临床数据不适宜获得)
二、一个自查清单
把以上14条浓缩成一句话:"NB要的不是完美的文档,而是完整的、一致的和可验证的文档。"
在你提交TD之前,花30分钟过一遍这个自查清单:
Intended Purpose在所有文档中完全一致? GSPR矩阵中每个条款都有"适用/不适用+理由"? 风险控制措施的有效性有验证证据? 所有交叉引用(报告号、版本号、日期)一致? 灭菌验证/再处理验证覆盖了最大复用次数? 生物相容性测试实验室证书在测试时点有效? 软件训练集和测试集已隔离? 网络安全文档包含了威胁建模方法? PSUR做了数据趋势分析和受益-风险结论? PMS计划有主动性活动(不止等投诉)? 水的质量标准有明确引用? 所有PDF文件≤100MB,有书签、可搜索? Equivalent器械声明有合同支持?
写在最后
这份Team-NB BPG V4最大的价值,不是它的92页内容,而是它传递的一个信号——
NB正在从"你写什么我审什么"走向"我告诉你我期待什么"。
对中国制造商来说,这不是坏消息。过去MDR的模糊地带,让很多企业在提交TD时"踩坑"而不自知。现在NB自己把扣分项摆在了台面上。
你需要做的就是——对照这份指南,检查你的技术文档。
信息来源
本文基于Team-NB Position Paper “Best Practice Guidance for the Submission of Technical Documentation under Annex II and III of MDR (EU) 2017/745” V4 (2026-04-21) 编写。全文92页,由20+家公告机构联合编制。
壹领医疗 Compliance Solutions for Medical Device
为医疗器械出海企业提供法规合规与验证服务
本文仅代表作者观点,具体法规适用请咨询专业法规顾问。

