 夜雨聆风
夜雨聆风随着全基因组测序(WGS)成本持续下降,越来越多的科研和临床项目开始大规模应用 WGS 数据。而在 WGS 数据分析中,拷贝数变异(Copy Number Variation,CNV)检测是一个绕不开的核心步骤——无论是肿瘤基因组学、罕见遗传病诊断,还是群体遗传学研究,都离不开 CNV 分析。
今天给大家分享一个非常实用的 AI Skill——cnv-caller-plotter,它可以帮助我们从 WGS 数据中自动检测 CNV,并生成高质量的全基因组可视化图谱,真正做到"一键从数据到图"。
一、cnv-caller-plotter 是什么?
cnv-caller-plotter 是由 AIPOCH 开发的一个开源 AI Skill,目前已有 583 颗 Star,专为全基因组测序数据的 CNV 分析设计。
核心功能包括:
CNV 检测:从比对好的测序数据(BAM 文件)中识别拷贝数增益(扩增)和缺失 基因组分箱(Segmentation):将基因组划分为等大小的区间(bin),用于拷贝数估算 多格式输入支持:兼容 BAM、VCF 等标准基因组学格式 发表级可视化:生成 PNG、PDF、SVG 格式的全基因组 CNV 图谱 标准化输出:以 BED 格式导出 CNV 结果,方便下游分析
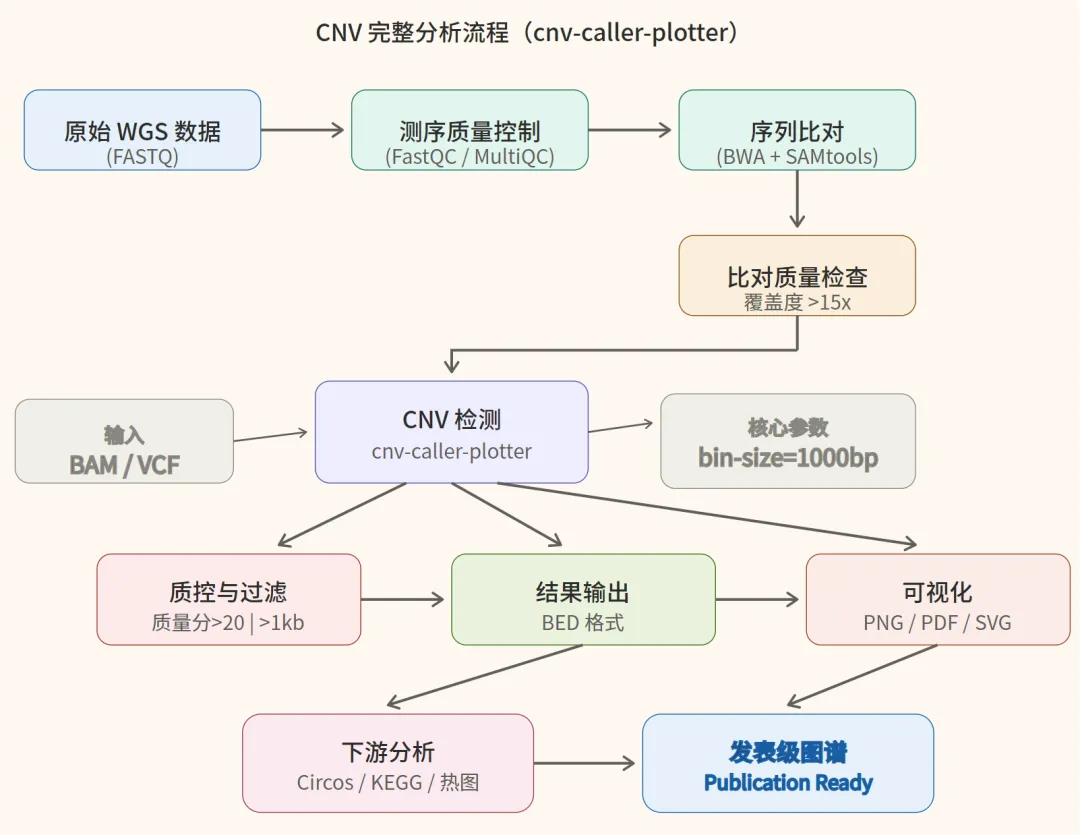
二、CNV 检测的核心原理
首先,我们来了解一下该工具是如何检测 CNV 的。
基于读深度(Read Depth)的检测策略
最核心的方法是读深度分析:将基因组切分成固定大小的"窗口"(bin),统计每个窗口内比对上的 reads 数量。正常二倍体区域的 reads 覆盖深度是均一的,而发生拷贝数扩增的区域覆盖会异常升高,发生缺失的区域覆盖则会降低。
from scripts.main import CNVCaller# 初始化 CNV 检测器,设定 bin 大小为 1000bpcaller = CNVCaller(bin_size=1000)# 从 BAM 文件中检测 CNVcnv_calls = caller.call_cnvs( input_file="sample.bam", reference="hg38.fa")# 查看检测结果for cnv in cnv_calls: print(f"{cnv['chrom']}:{cnv['start']}-{cnv['end']}")if cnv['cn'] > 2: print(f" 类型:扩增(拷贝数 = {cnv['cn']})")elif cnv['cn'] < 2: print(f" 类型:缺失(拷贝数 = {cnv['cn']})")Bin 大小怎么选?
一句话总结:覆盖度越高,bin 可以越小,检测分辨率越高。
三、三大应用场景
场景一:肿瘤基因组分析(肿瘤-正常对)
这是最常见的应用场景。肿瘤样本中存在大量体细胞拷贝数变异(SCNA),通过与配对正常组织对比,可以精准识别肿瘤特异性的扩增和缺失。
工作流程如下:
对肿瘤和正常 BAM 文件分别进行 CNV 检测 两组结果对比,过滤掉正常样本中已存在的胚系 CNV 保留肿瘤特有的体细胞变异 注释癌症驱动基因(如 MYC 扩增、TP53 缺失) 生成发表级图谱
一个典型的输出示例:
体细胞 CNV 汇总: 总变异数:47 处 扩增:12 处(包含 MYC、EGFR) 缺失:35 处(包含 TP53、PTEN)关键变异: chr8:128000000-129000000 CN=8(MYC 高度扩增) chr17:7000000-8000000 CN=0(TP53 纯合缺失)场景二:罕见遗传病 CNV 诊断
对于疑似基因组病的患者,高灵敏度 CNV 检测至关重要。此场景需要:
使用更小的 bin(约 500bp)以提高灵敏度 过滤人群中常见 CNV(DGV、gnomAD 数据库) 重点关注低频(<1%)的罕见 CNV 与 OMIM、ClinVar、DECIPHER 等数据库交叉注释
场景三:群体遗传学研究
当需要分析大批量样本时,该工具支持批量处理,可用于:
识别在特定疾病群体中反复出现的 CNV 热点区域 进行 CNV 负荷(burden)分析 病例-对照关联检验
四、快速上手:从数据到图只需几步
环境准备
# 需要 Python 3.10+,安装必要依赖pip install pysam numpy matplotlib pandas# 创建参考基因组索引samtools faidx hg38.fa# 确保 BAM 文件已建索引samtools index sample.bam运行分析
# 基本用法:输入 BAM,输出 CNV 结果python scripts/main.py \ --input sample.bam \ --reference hg38.fa \ --output ./cnv_results \ --bin-size 1000 \ --plot-format pdf输出文件结构
分析完成后,输出目录结构如下:
cnv_results/├── cnv_calls.bed # CNV 结果(BED 格式)├── cnv_plot.pdf # 全基因组 CNV 可视化图谱└── analysis_summary.json # 统计汇总信息五、与其他工具的联动
cnv-caller-plotter 并不是孤立使用的,它有明确的上下游工具衔接:
上游(数据预处理):
fastqc-report-interpreter:评估测序质量alignment-quality-checker:检查 BAM 文件覆盖均一性
下游(结果深挖):
circos-plot-generator:生成染色体环形图go-kegg-enrichment:对 CNV 区域内的基因做功能富集分析heatmap-beautifier:多样本 CNV 热图可视化
完整流程:
原始 WGS 数据 → 质控 → 比对 → cnv-caller-plotter → 功能注释 → 发表图谱六、几个常见坑,提前避开
坑 1:覆盖度不足WGS CNV 检测至少需要 15-20x 的覆盖度,低于这个阈值假阳性率会急剧升高。
坑 2:参考基因组版本不一致BAM 文件比对用的是 hg19,但 CNV 分析用了 hg38 的参考,结果坐标会乱套。务必确保全程用同一个版本。
坑 3:肿瘤纯度问题肿瘤样本中混有大量正常细胞时,CNV 信号会被"稀释"。纯度低于 20% 时,部分 CNV 可能检测不到。
坑 4:不过滤重复区域着丝粒、端粒、节段重复区域的 reads 比对不可靠,会产生大量假阳性 CNV。务必在后处理步骤中过滤这些区域。
小结
cnv-caller-plotter 是一个功能完善、使用简便的 CNV 分析 AI Skill,覆盖从数据输入、CNV 检测、质控过滤到可视化输出的完整流程,适用于肿瘤基因组学、罕见遗传病以及群体遗传学等多个场景。
CNV 分析的工具和方法有很多,大家可以根据自己的需求和数据类型灵活选择。如果你正在使用 WGS 数据并希望快速获得高质量的 CNV 结果,不妨试试这个工具,欢迎在评论区分享你的使用体验!
文中有表述不准确的地方,也希望大家不吝指正 😊
参考资料
cnv-caller-plotter GitHub 仓库:https://github.com/aipoch/medical-research-skills Skill 详情页:https://skillsmp.com/zh/skills/aipoch-medical-research-skills-scientific-skills-data-analysis-cnv-caller-plotter-skill-md Database of Genomic Variants (DGV):http://dgv.tcag.ca gnomAD Structural Variants:https://gnomad.broadinstitute.org ClinVar:https://www.ncbi.nlm.nih.gov/clinvar DECIPHER:https://www.deciphergenomics.org
