 夜雨聆风
夜雨聆风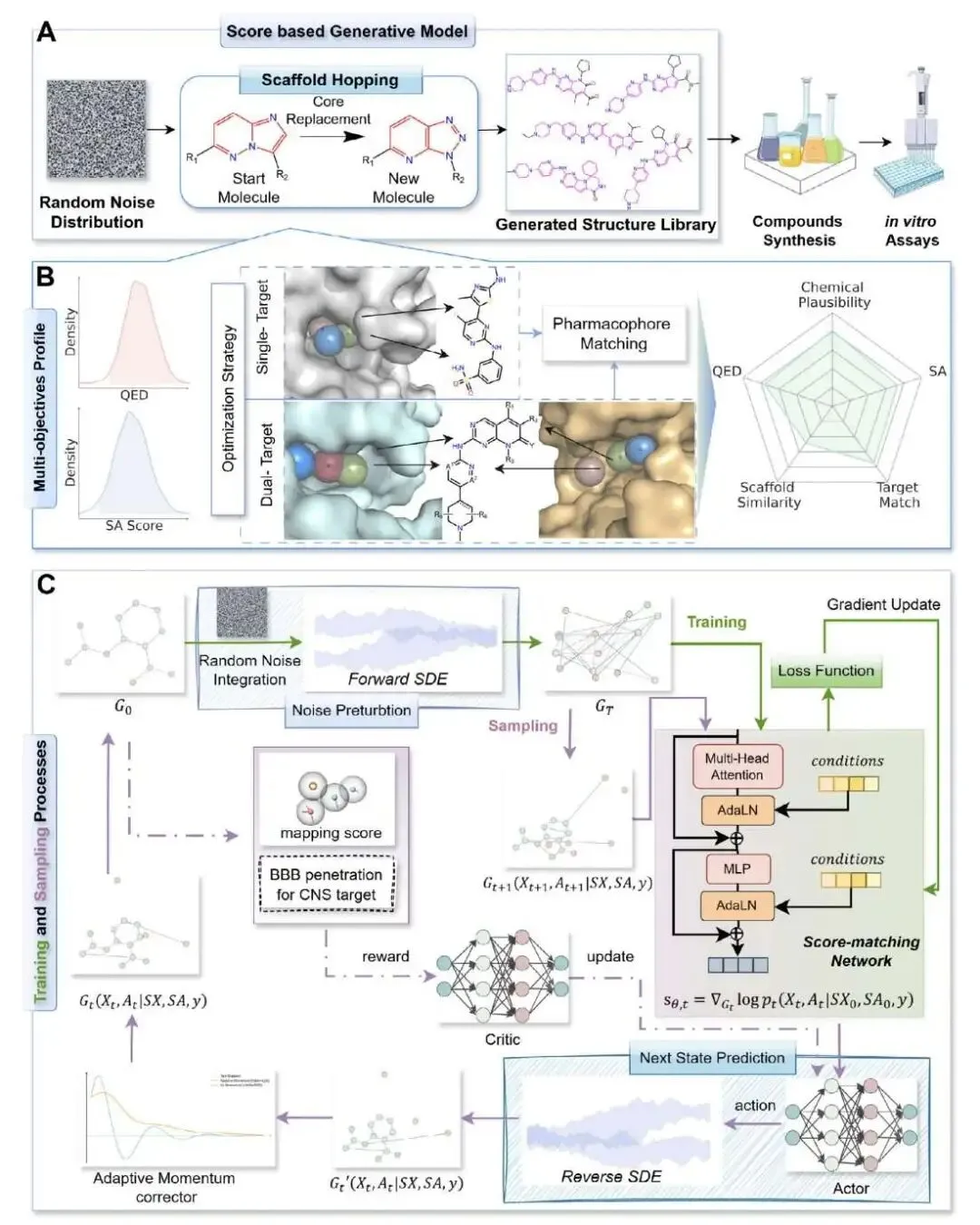
<<< 左右滑动见更多 >>>
榴莲忘返AIDD
供稿 | 别开枪是我
审稿 | 吉星
目录
SMarT-Diff,一个基于扩散模型的 AI 框架,通过可控的骨架跳跃策略,成功设计并验证了活性优于阳性对照的纳摩尔级 LRRK2 抑制剂。 ProteoCast 通过为每个蛋白定制评估标准和挖掘无序区功能,能更准确地预测突变影响,并已通过 CRISPR 实验验证。 LAffAb 是一种计算设计策略,它通过预先筛选出不破坏结构稳定性的「安全」突变位点,从而能够一次性构建出同时优化亲和力和稳定性的高质量抗体组合库。 ProtSpace 是一个完全在浏览器中运行的可视化工具,它让探索数十万蛋白质基于语言模型的结构 - 功能关系变得前所未有的直观和便捷。 Meta-encoder 提供了一个即插即用的框架,通过融合多个现有病理 AI 模型的特征,无需重新训练即可获得更稳健、更强大的癌症检测与分子预测能力。

1. AI 骨架跳跃新范式:生成 nM 级 LRRK2 抑制剂
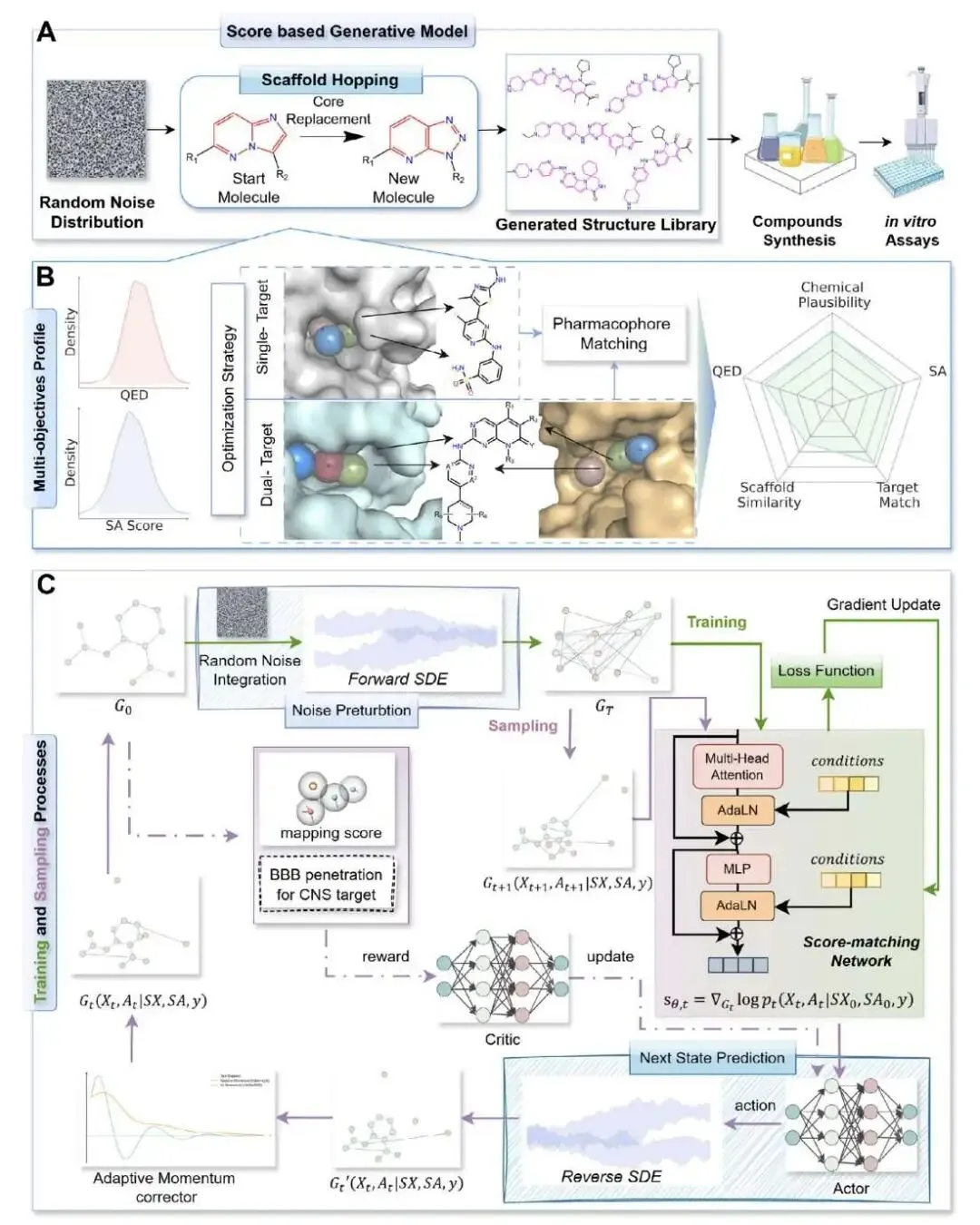
做先导化合物优化(Lead Optimization)时,我们总是在一个矛盾里挣扎:要么在已知的骨架上小修小补,稳妥但很难有突破;要么就大胆尝试全新的骨架,也就是骨架跳跃(scaffold hopping),风险高但可能带来惊喜。大部分生成式 AI 模型其实更擅长前者,生成的分子看起来新,但骨架往往和训练集里的差不多。
SMarT-Diff 这个新模型似乎找到了一个平衡点。
最打动人的地方,是他们不只发了篇算法文章,还直接下场做了湿实验。研究者们针对 LRRK2 靶点,从模型生成的分子里挑了三个去合成、测试。结果,最好的一个分子 lrrk2_m_1001 的 IC50 值做到了 1.544 nM。这个数据很亮眼,因为它比阳性对照药 LRRK2-IN-1(IC50 = 3.141 nM)的活性还要高出一倍。对于一个从头生成的全新分子来说,这是个非常扎实的成果。
那么,它是怎么做到的?
SMarT-Diff 的核心是一个基于图扩散 Transformer 的生成模型。你可以把它想象成一个「分子工匠」。我们先给它一个 Bemis–Murcko 骨架作为起点,告诉它:「这是我们大概想要的样子」。但同时,我们又给它设定了几个目标:生成的分子类药性(QED)要好,合成起来不能太难(SA),还要符合我们指定的药效团。
最有意思的是它的采样过程,分了两步走。
第一步,一个内部的预测 - 校正循环(RA)负责稳定地生成化学上合理的分子,保证基本的「化学语法」不出错。这一步倾向于生成与初始骨架比较相似的分子。
第二步,引入了一个外部的强化学习循环(A2C)。这一步是实现「跳跃」的关键。它会根据药效团匹配度给模型奖励,但如果生成的分子骨架和初始骨架太像,它就会给出「惩罚」。这样一推一拉,模型就被迫去探索那些既能满足药效团要求、又具有全新骨架的化学空间。
通过调节这个「惩罚」力度,研究者能精确控制骨架跳跃的幅度。实验数据显示,在不加 A2C 控制时,生成分子的骨架相似度高达 0.73,基本就是在做微调。加上 A2C 后,相似度可以被拉低到 0.36-0.41 的区间。对于药物化学家来说,0.4 左右的相似度,通常就意味着一个有意义的、值得探索的新骨架了。
当然,光有新骨架还不够,活性得保住。研究者们检查了那些相似度低于 0.5 的新分子,发现它们的对接分数(Glide SP)依然很强,中位数在 -8.57 kcal/mol 左右。这说明 SMarT-Diff 学到的是结合口袋所需的关键相互作用模式(药效团),而不是简单地复制某个特定骨架的形状。
为了证明自己的实力,SMarT-Diff 还和 PMDM、DECOMPOPT 等好几个主流的骨架跳跃模型同台竞技。结果显示,它在有效性、新颖性、独特性、多样性和各项成药性指标上取得了最佳的综合平衡,特别是在「成功率」这项综合指标上排名第一。
最后,这个框架的适用性也很广。研究者把它用在一个 GPCR 靶点 GLP-1R 上,不用重新训练模型就能得到不错的结果。他们甚至还尝试了 GSK3β/JNK3 双靶点药物设计,同样取得了积极的计算化学验证结果。这表明 SMarT-Diff 的底层逻辑具有很好的普适性,不局限于某一类特定靶点。
📜Title: Diffusion-based generative model with scaffold-hopping strategy yields highly potent bioactive molecules
🌐Paper: https://doi.org/10.1002/advs.75674

2. ProteoCast: 预测突变不再靠猜,看进化怎么说
在药物研发和基因组学里,我们总会遇到一个头疼的问题:一个氨基酸突变,究竟会产生多大影响?是无关痛痒,还是会引发疾病?预测工具很多,但总感觉隔靴搔痒。最近 Nature Communications 上发表的 ProteoCast 工作,提供了一个相当漂亮的解法。
这套方法的思路是:让进化本身来告诉我们答案。如果一个氨基酸位点在数百万年的进化中几乎没变过,那任何对它的改动都可能带来大麻烦。
具体他们是怎么做的?
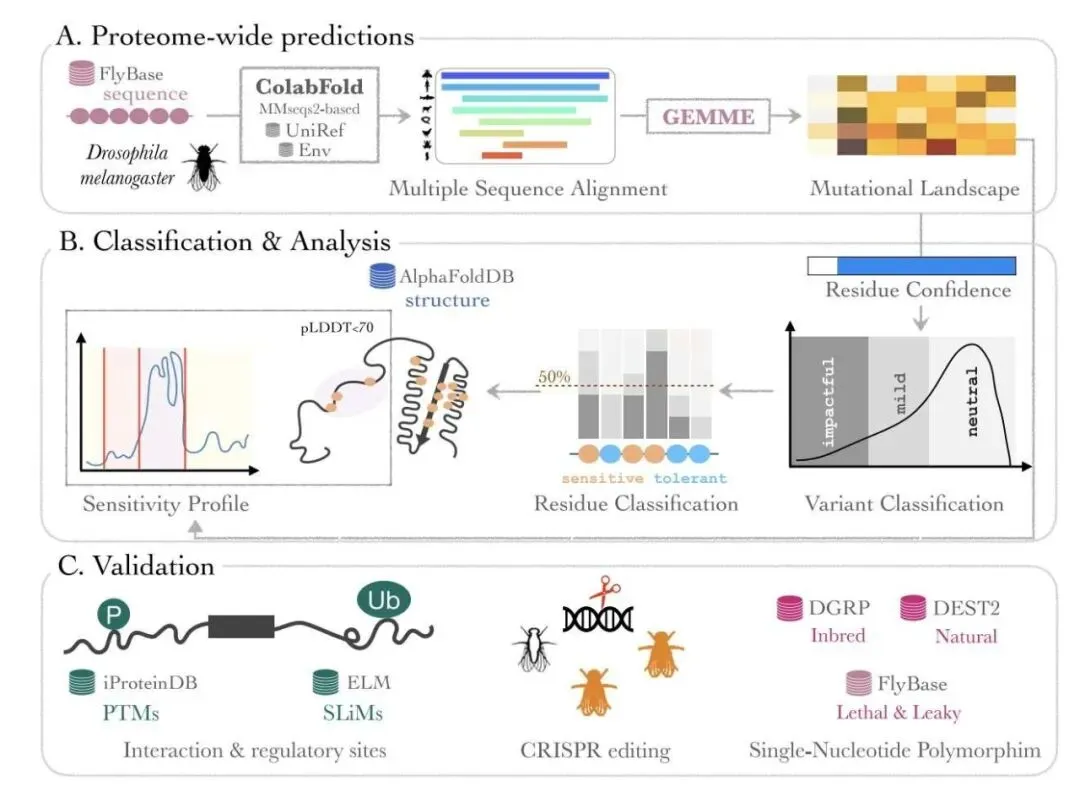
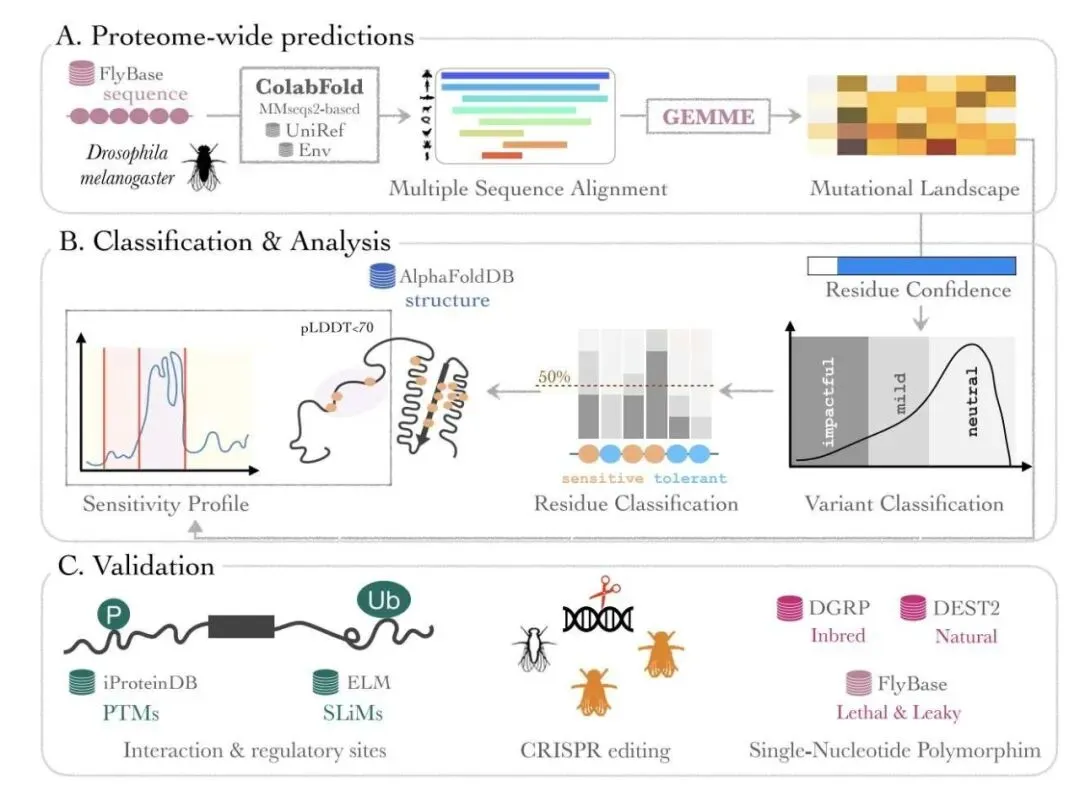
第一步,收集数据。研究者利用 ColabFold 对果蝇的全部蛋白质组进行了多序列比对(Multiple Sequence Alignments, MSAs)。这就好比是找到了每个蛋白在不同物种中的所有「亲戚」,汇编成了一部厚厚的家族史。
第二步,计算突变影响。他们用一个叫 GEMME 的工具,基于这份家族史,为每个蛋白的每个位点上可能发生的 19 种氨基酸替换都打了一个分。分数越差,说明这个突变在进化中越不被容忍,也就越可能是有害的。到这里,很多类似工具的工作就结束了。
但 ProteoCast 的真正亮点在后面。
研究者意识到,用一个统一的「有害」分数线去衡量所有蛋白,是不科学的。有些蛋白是「保守派」,任何风吹草动都受不了;有些则是「激进派」,结构灵活,对突变容忍度高。
所以他们引入了一个巧妙的创新:为每个蛋白建立自己独特的评估体系。他们对每个蛋白的所有突变分数分布,拟合了一个三成分高斯混合模型(Gaussian Mixture Model)。这个模型能自动把分数划分为三类:中性(neutral)、轻微影响(mild)和显著影响(impactful)。这样一来,每个蛋白都有了自己专属的「及格线」,判断突变影响自然就更准了。
另一个让人印象深刻的地方,是它对无序蛋白区的处理。很多蛋白区域没有固定的三维结构,像一根晃来晃去的绳子。传统方法很难分析它们,但这些区域往往藏着重要的调控位点,比如磷酸化位点或短线性基序(Short Linear Motifs, SLiMs)。
ProteoCast 并不依赖三维结构。它直接在蛋白序列这条一维线上分析突变敏感性分数。通过一种叫做变点检测(changepoint detection)的算法,它可以自动找出那些对突变「特别敏感」的片段,也就是功能「岛屿」。结果发现,这些「岛屿」和已知的磷酸化位点、短线性基序高度重合。这意味着,我们终于有了一个可靠的工具,去系统性地发掘无序区的秘密。
当然,计算说得再好,也得实验来验证。研究者用 CRISPR 技术在果蝇体内验证了他们的预测。他们挑选了一个关键的代谢酶 Naprt,根据 ProteoCast 的预测,引入了三种「显著影响」突变和两种「中性」突变。
结果非常干净利落:携带「显著影响」突变的果蝇无法存活到纯合子阶段,符合隐性致死的表型;而携带「中性」突变的果蝇则能正常发育为纯合子。这直接证明了 ProteoCast 预测的可靠性。
这项工作不仅提供了一个强大的工具,更重要的是提供了一种思考方式。它告诉我们,进化数据里藏着远超我们想象的信息,而如何解读这些信息,决定了我们能走多远。对于靶点发现、致病机理研究,甚至是蛋白质工程,ProteoCast 都打开了新的可能。
📜Title: Proteome-wide prediction of the functional impact of missense variants with ProteoCast
🌐Paper: https://doi.org/10.1038/s41467-024-48671-1
💻Code: https://hub.docker.com/r/marinaabakarova/proteocast/

3. 别再迭代了:计算如何「一步到位」优化抗体?
做抗体优化的人都有过这种头疼的经历:费了九牛二虎之力,通过几轮迭代把亲和力提上去了,结果一测稳定性,发现蛋白变得极不稳定,或者表达量一落千丈。这就像玩打地鼠游戏,按下葫芦浮起瓢,让人筋疲力尽。
如果我们能有一种方法,在设计阶段就同时考虑亲和力和稳定性,一步到位地构建一个富含高质量候选分子的库,那会怎么样?
来自 Weizmann 科学研究所 Fleishman 实验室的这篇工作就提出了这样一种策略,他们称之为 LAffAb (Libraries of Affined Antibodies)。这个方法的核心思路非常直观:在尝试引入新的、能增强结合的突变之前,先弄清楚哪些位置的突变会把整个结构搞垮。
先排除「雷区」,再寻找「宝藏」
抗体的 CDR (互补决定区) 区就像一台精密调谐的仪器。你不能随便更换零件,否则整个机器都会罢工。LAffAb 的第一步,就是用 Rosetta 软件对 CDR 区进行一次全面的「安全检查」。
这个过程有点像对每个氨基酸残基进行压力测试。研究者们通过计算(比如丙氨酸扫描和氢键分析)来识别那些对维持 CDR 骨架构象、或者稳定关键的埋藏氢键网络至关重要的残基。这些位置就是「雷区」,会被标记为「不可触摸」。
剩下的那些位置,就是「安全突变区」。在这些安全区里,研究者们再结合抗体序列数据库的统计信息(比如 PSSM),挑选那些在天然抗体中出现频率较高的氨基酸作为候选突变。这样做的目的是保证引入的突变是「符合自然规律」的,进一步降低风险。
「一劳永逸」的组合库设计
这种「先避险,后寻宝」的策略最大的好处在于,它能有效降低负向上位性(negative epistasis)的风险。所谓负向上位性,就是指两个单独看都是有益的突变,组合在一起后却产生了意想不到的坏效果。通过预先筛选出那些不影响核心结构稳定性的突变,它们被组合在一起时互相「打架」的概率就大大降低了。
这使得研究者可以大胆地跳过缓慢的单点突变迭代,直接设计一个包含多个突变组合的大型文库。这是一种「一步到位」的策略,旨在让文库从一开始就富含功能和稳定性俱佳的分子。
实验结果怎么样?
我们来看几个例子。
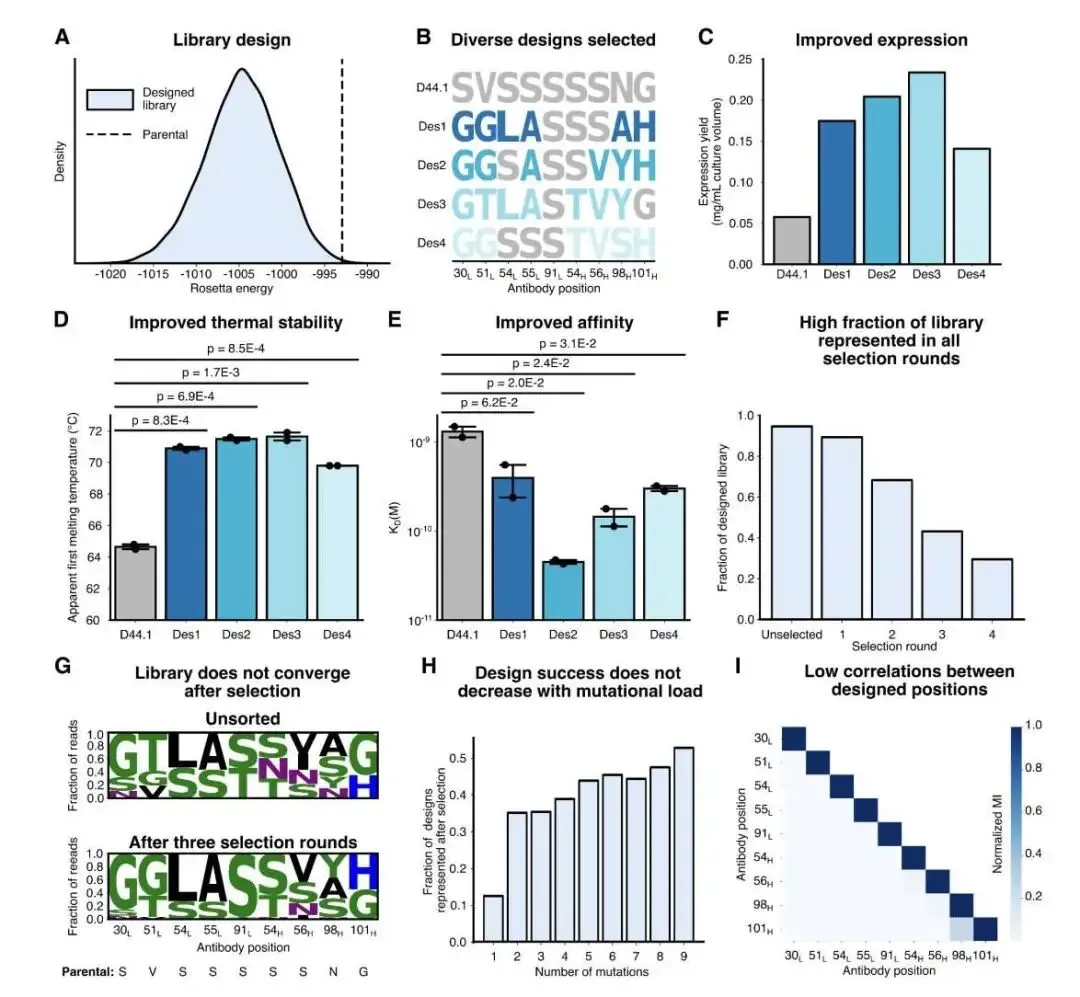
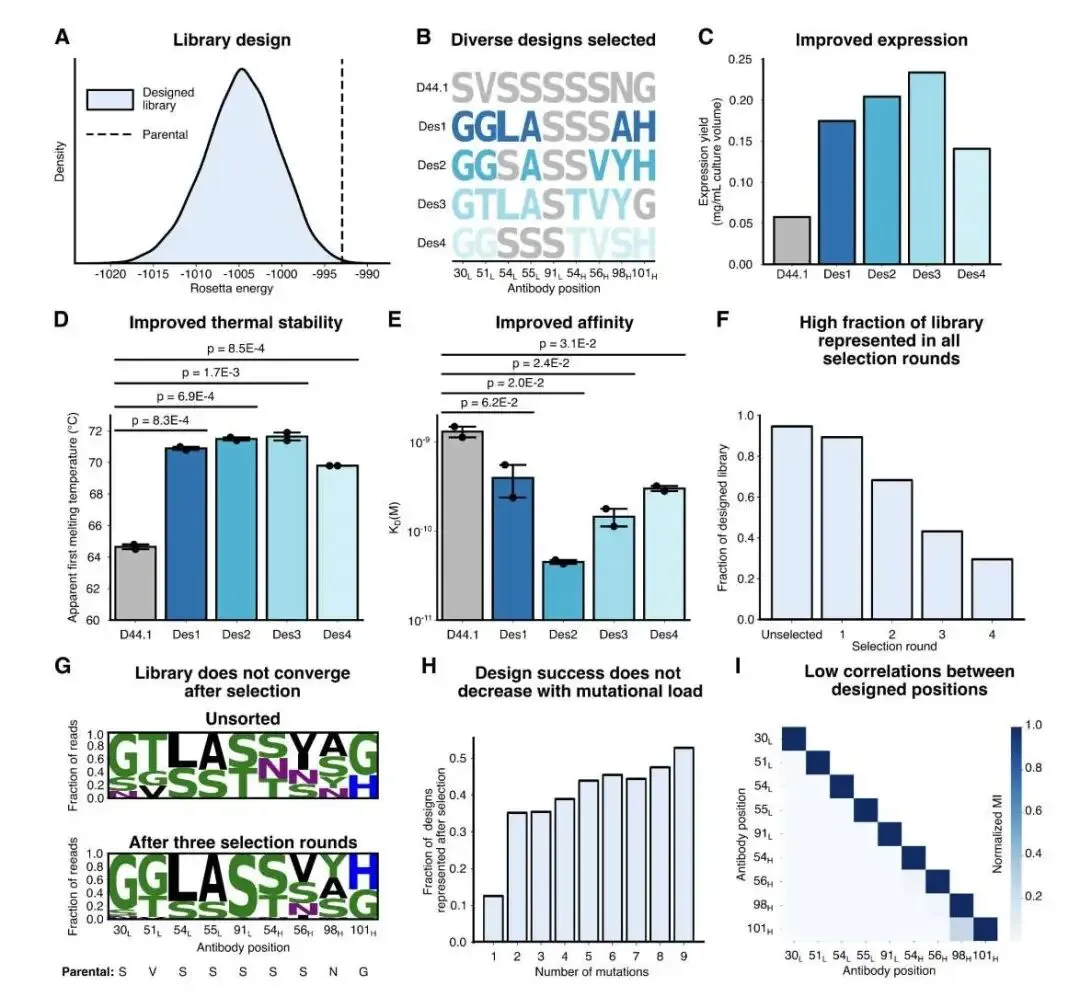
首先是经典的 D44.1 抗体,它与溶菌酶(HEWL)结合。计算显示,在 59 个 CDR 残基中,只有 18 个被认为是「安全可突变」的。一个有意思的发现是,这些安全位点大多不直接参与抗原结合。这说明,提升亲和力不一定非要增加新的结合作用力,通过稳定 CDR 的构象,使其「预组织」成更适合结合的状态,同样能达到目的。
基于这个结果,他们构建了一个包含近 7000 个变体的组合库。经过噬菌体展示筛选后,筛选出的优势克隆普遍带有 4-8 个突变。其中最好的一个设计,亲和力从亲本的 5 nM 提升到了 50 pM,足足高了 30 多倍。更关键的是,这个分子的 CHO 细胞表达量和热稳定性也得到了同步提升,真正实现了一石二鸟。深度测序数据也证实,文库的多样性很好,没有迅速坍缩到某一个最优解上,印证了其设计理念的成功。
接下来是一个更具挑战性的治疗性抗体案例:6G08。它的靶点是 FcγRIIb,但必须避开与之序列高度同源(胞外域 93% 一致)的 FcγRIIa,以保证特异性。研究者仅设计了 10 个各带 6 个突变的变体,就从中找到了 2 个分子,不仅对 FcγRIIb 的亲和力提升了近 30 倍,同时对 FcγRIIa 的结合非常弱,成功维持了特异性。
他们还挑战了一个已知的「问题分子」——Urelumab。这个抗体有聚集和自缔合的问题,成药性不佳。这次的目标是改善成药性,同时保持亲和力不变。结果显示,基于 LAffAb 的低能量设计确实表现出更好的热稳定性和更低的聚集倾向,其中 6 个设计在成药性改善的同时,亲和力与亲本基本持平。
方法的边界在哪里?
当然,LAffAb 也不是万能的。它的成功严重依赖于一个高质量的抗原 - 抗体复合物晶体结构。
研究者们坦诚地分享了一个失败案例——西妥昔单抗(Cetuximab)。他们的设计没有成功。事后分析发现,他们所使用的晶体结构中,缺失了几个在结合界面起到「桥梁」作用的关键水分子。由于计算模型没有「看到」这些由水介导的相互作用,它给出的「安全区地图」就是错误的。这给我们提了个醒:任何计算设计的起点都是高质量的实验数据,输入的是垃圾,输出的也只能是垃圾。
LAffAb 提供了一种非常抗体工程新范式。它不是要取代实验,而是让实验变得更高效。通过将结构生物学、能量计算和序列分析巧妙地结合,它为我们指明了一条如何从源头上就设计出更优秀的抗体库的道路,从而大大节省后续筛选和优化的时间和成本。
📜Title: Energy-guided combinatorial co-optimization of antibody affinity and stability
🌐Paper: https://www.biorxiv.org/content/10.1101/2025.11.26.690765
💻Code: https://github.com/Fleishman-Lab/LAffAb_public

4. ProtSpace:在浏览器里探索蛋白质功能宇宙
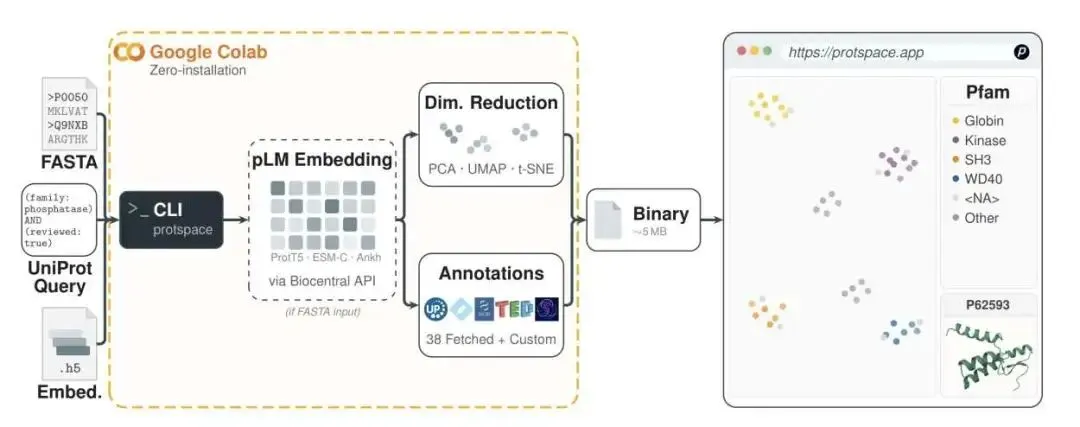
做蛋白质研究,我们绕不开一个经典工具:BLAST。输入一个序列,它会告诉你哪些蛋白质和它「长得像」。这个方法很管用,但也有其局限。它就像是通过拼写来查字典,如果两个词拼写差异很大,即使意思相近,你也找不到。在蛋白质的世界里,这意味着如果两个蛋白序列相似性不高,即使它们功能相同、结构类似,BLAST 也可能无能为力。
蛋白质语言模型(pLM)的出现改变了这一点。它不再关注序列的「拼写」,而是学习序列背后蕴含的生物学「含义」。通过 pLM 得到的嵌入向量 (embedding),能把功能或结构相似的蛋白质在数学空间中拉近。这是一个巨大的进步,但对很多湿实验科学家来说,使用这些 embedding 并不轻松,通常需要编程和强大的计算资源。
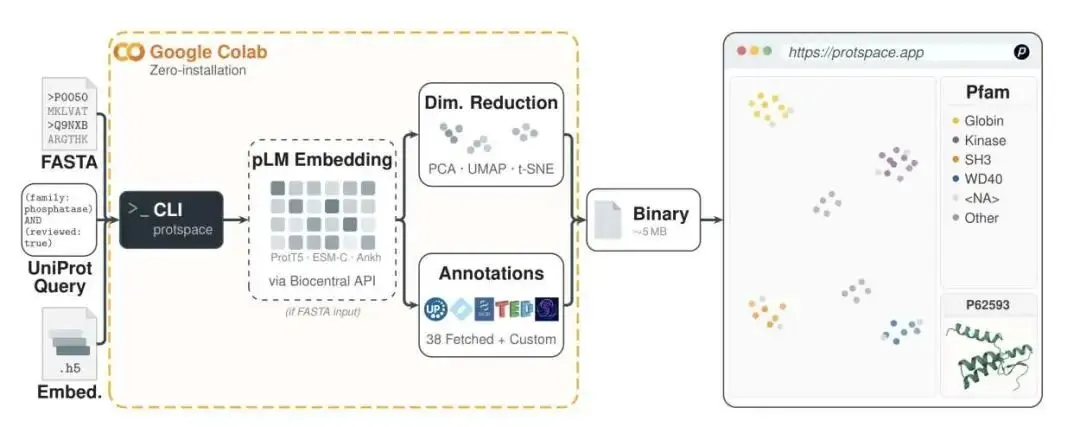
ProtSpace 这个工具把整个流程搬到了你的浏览器里。
它的核心理念很简单:让任何人都能像逛地图一样,直观地探索 pLM 构成的蛋白质宇宙。研究者们把整个 Swiss-Prot 数据库(包含约 57.3 万条蛋白质)的 ProtT5 嵌入向量投射到一个 2D 平面上。你不需要安装任何软件,也不用上传任何数据,打开网页就能用。
这是怎么做到的?背后是扎实的工程实现。它利用了 WebGL 加速渲染和四叉树(quadtree)空间索引技术。简单来说,即使屏幕上有几十万个数据点,你进行缩放、平移、悬停点击等操作,响应依然非常流畅。
从「序列相似」到「功能相近」的思维转变
ProtSpace 真正强大的地方,在于它提供了一种新的假设生成界面。
论文中有一个关于 β-内酰胺酶超家族的例子。这个家族的酶根据催化机制被分为 A、B、C、D 四个 Ambler 类别。如果用传统的序列相似性网络来分析,结果可能比较混乱。但在 ProtSpace 的 pLM 嵌入空间视图中,这四个类别形成了清晰的、可区分的集群。
这意味着,即使两个酶的序列差异很大,只要它们的催化机制(也就是核心功能)相似,pLM 就能捕捉到这一点,并将它们放在相近的位置。更有意思的是,研究者还发现了一些被错误注释的蛋白质——它们虽然被标记为某个类别,但在视图中却跑到了另一个类别的集群里。这为功能修正和发现新的蛋白家族提供了直接线索。
一个集大成的数据探索平台
ProtSpace 不只是一个漂亮的散点图。它的价值在于信息的整合。
丰富的注释系统:它自动从 UniProt、InterPro、NCBI 等多个权威数据库中抓取了 38 种注释信息。你可以一键切换,用不同的标签(如 GO 功能、EC 酶号、亚细胞定位、蛋白家族)给这些点上色,从不同维度观察蛋白质的分布规律。 创新的多标签可视化:一个蛋白质通常有多个功能域(domain)。传统的视图只能用一种颜色标记,信息损失很大。ProtSpace 用饼图来表示每个点,一个饼图的不同扇区可以代表不同的功能域。这样一来,你就能一眼看出某个区域的蛋白质是不是普遍具有相似的「功能域架构」。 整合 3D 结构:这是我最喜欢的功能。当你在 2D 视图中发现一个感兴趣的蛋白质,点击它,旁边就会弹出一个 Mol* 浏览器,直接加载它在 AlphaFold 数据库里的预测结构。这个功能打通了从宏观功能聚类到微观三维结构的探索链路。你可以快速验证:这个集群里的蛋白质,它们的活性口袋在结构上是否也相似?
对于药物研发来说,这个工具的想象空间很大。无论是寻找新靶点、分析靶点家族成员、还是预测潜在的脱靶效应,这种基于「功能」而非「序列」的全局视图,都可能带来全新的发现。它把强大的 pLM 分析能力,从少数计算生物学家的终端里解放出来,变成了一个人人都能上手的网页应用。
📜Title: ProtSpace: Protein Universe in Your Browser
🌐Paper: https://www.biorxiv.org/content/10.64898/2026.05.04.722720
💻Code: https://github.com/tsenoner/protspace_web

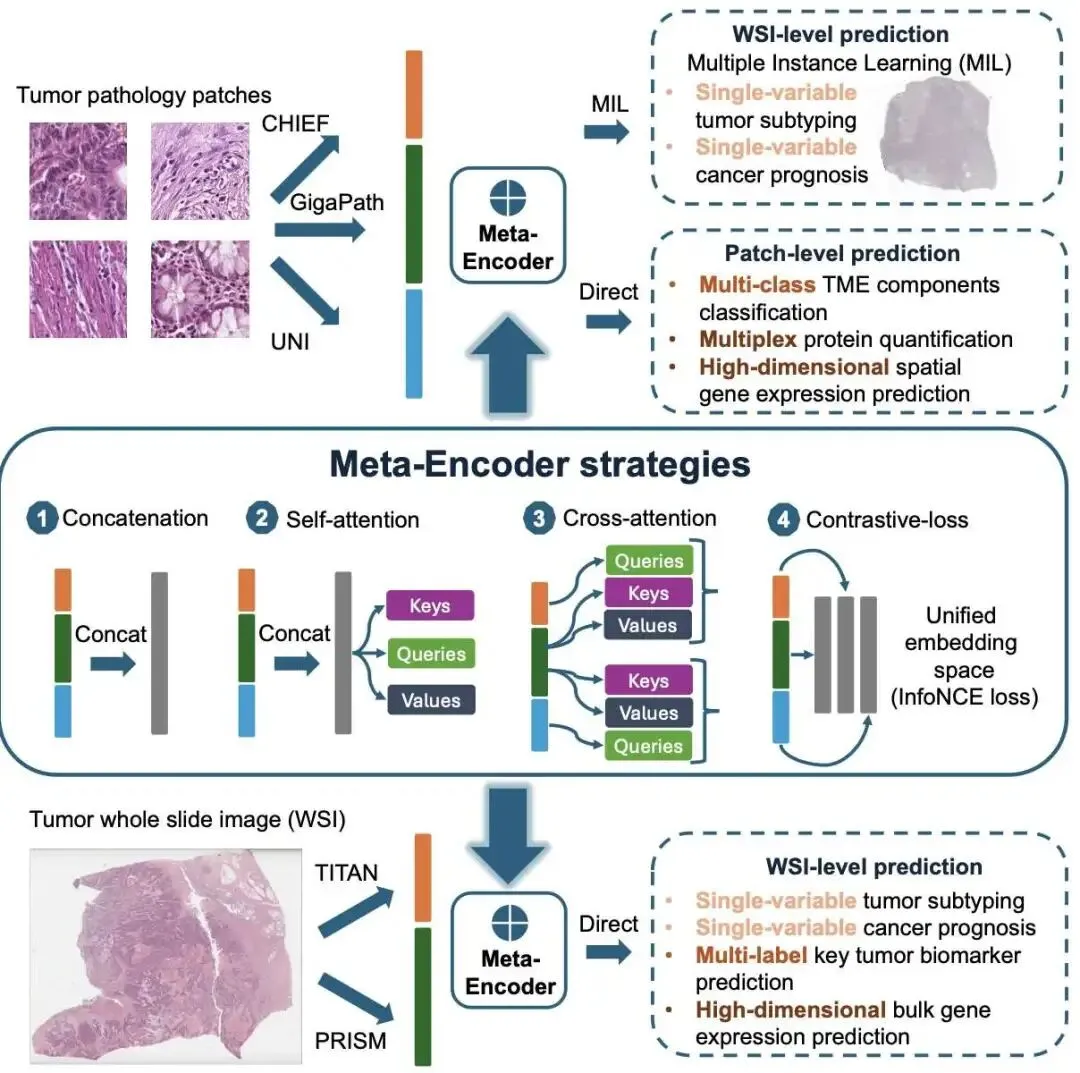
5. 病理 AI 模型太多怎么选?Meta-encoder:我全都要!
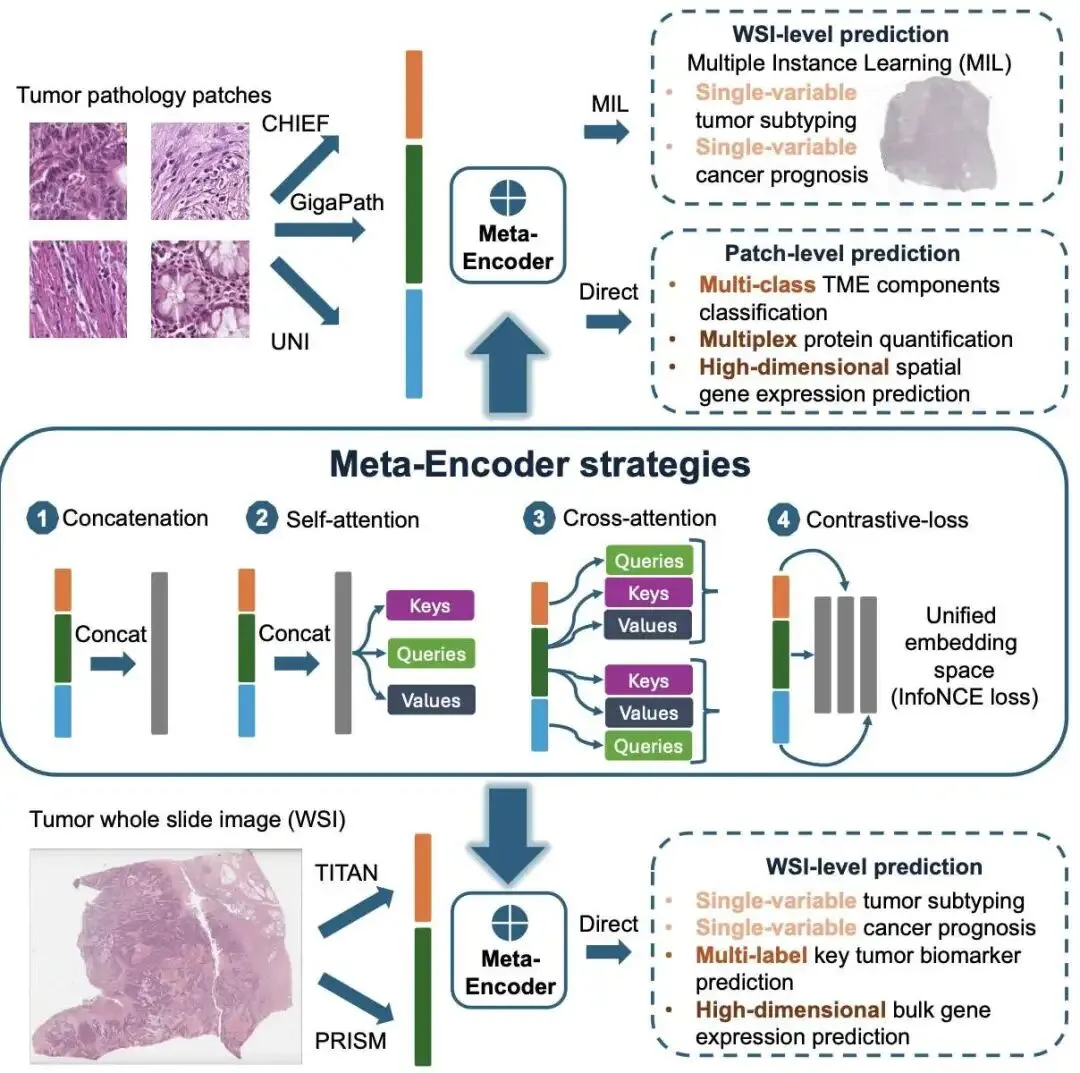
在计算病理领域,我们正面临一个「幸福的烦恼」:优秀的病理学基础模型(Foundation Models)越来越多,比如 CHIEF、GigaPath、TITAN 等。它们就像一整套高度专业化的工具,每个都有自己的长处。问题来了,面对一个新的研发任务,我们该选哪个模型?选错了,就意味着浪费宝贵的计算资源和时间。更头疼的是,由于数据隐私和模型架构各异,想把这些模型拿来从头整合训练,几乎是不可能的。
这篇论文提出的 Meta-encoder 框架,就是来解决这个问题的。它的思路是:既然不能重新训练,那我们能不能造一个「万能转接头」,把这些现成模型的输出(也就是特征嵌入)直接拼起来用?
这是它的工作原理
研究者们设计了这个即插即用的 Meta-encoder。它直接拿各个预训练好的基础模型的输出结果,在下游任务微调时进行融合。这样做的好处是,我们根本不需要接触到原始的预训练数据,也不用去动那些复杂的模型内部结构。
作者测试了四种融合策略:
直接拼接:最简单粗暴的方法,把不同模型的特征向量首尾相连。就像把几份不同的报告用订书机钉在一起。 拼接 + 自注意力(Self-attention):这是个更办法。拼接后,加一个轻量级的自注意力层。它好比一个项目经理,能快速浏览这份「合并报告」,并判断出针对当前任务,哪一部分信息更关键,然后给它更高的权重。 交叉注意力(Cross-attention):让不同模型的特征互相「交流和参考」。 对比损失正则化:在微调时,强制让不同模型的视角趋于一致。
不同任务,不同策略
实验结果揭示了一个清晰的模式。
对于相对简单的任务,比如肿瘤亚型分类或生存风险预测,最简单的「直接拼接」策略效果就很好,几乎能达到单一最佳模型的水平。这让它成了一个非常实用的「安全选项」。当你不知道该用哪个模型时,把它们都用上然后拼起来,结果基本不会差。
一个有意思的发现是,在全切片图像(WSI)分型任务中,使用自注意力机制虽然对 AUC(Area Under the Curve)提升不大,但显著改善了模型的概率校准。这意味着,模型给出的「置信度」变得更可靠了。在临床应用中,一个模型不仅要「猜得对」,更要「知道自己有多大把握猜对」,这一点至关重要。
复杂任务才是它的主场
Meta-encoder 真正的威力体现在结构化、高维度的分子预测任务上。比如,同时预测多种生物标志物(如 RAS/BRAF/MSI 状态)、定量多种蛋白表达水平,或者预测空间基因表达。在这些场景下,任何单一模型都可能存在「视野盲区」。
而基于注意力的融合方法,特别是自注意力,在这里大放异彩。它能显著超越任何一个单一模型。在一个独立的结直肠癌数据集上进行外部验证时,自注意力融合模型将平均 AUC 从最佳单一模型的 0.656 提升到了 0.737。更关键的是,在固定 90% 特异性的前提下,它的灵敏度从 35.9% 飙升至 60.8%。这种在全新数据上的表现,充分证明了其强大的泛化能力和鲁棒性。
它为什么有效?
研究者通过 SHAP 分析来探究其内部机制。结果表明,Meta-encoder 不是简单地把参数堆砌起来,而是像一个指挥家。在预测某个特定蛋白时,它可能主要依赖 UNI 模型的特征;而在预测另一个蛋白时,则可能更侧重 GigaPath 的信息。它会根据具体的分子靶点,动态地调整从每个基础模型中提取信息的权重。这证实了核心的假设:不同的基础模型学到了互补的知识,而 Meta-encoder 成功地将这些知识整合了起来。
给研发同行的实践指南
这篇论文给出了非常清晰的部署建议:
这项工作为我们如何有效利用日益庞大的病理 AI 模型生态系统,提供了一个非常实用的范式。它将我们的思考从「我该选哪个模型?」转变为「如何让它们协同工作?」。
📜Title: Meta-encoder: a unified integration framework for multiple pathological foundation models in cancer detection
🌐Paper: https://doi.org/10.1038/s41467-026-71558-x
💻Code: Not available
