 夜雨聆风
夜雨聆风
写在前面
CHGNet、MatterSim、SevenNet跑通了 7 个电池材料结构的弛豫和 MD 短程测试。 LiC6 在CHGNet、MatterSim、SevenNet 的50步弛豫中都未收敛。 graphite 100 步MD在三个可运行模型里都出现严重温度飞升,不能作为稳定300 K MD使用。 Li metal 1000 步MD在CHGNet、MatterSim、SevenNet中都无NaN、未炸飞。 能弛豫不等于能稳定MD;通用势必须做体系内短程MD测试。 这类模型可以作为快速筛选工具,但不能直接替代DFT或严肃MD验证。
1. Introduction
现在大原子模型和通用机器学习势很火,但普通材料科研人真正关心的不是榜单第一名,而是几个很实际的问题:
我的结构能不能丢进去? relax会不会收敛? MD会不会温度飞升? 同一个模型在不同结构上是不是表现一致?
所以我们这次测评按这个思路做,看一看一个小时内我到底能不能搞定电池材料的模拟,我用那个MLIP。
2. Method
本次只展示有效进入能量/力计算并产生 relax 与 MD 结果的 CHGNet、MatterSim、SevenNet。
测试结构来自同一批 benchmark:Li metal、graphite、LiC6 toy、LiCoO2、LiCoO2 Li空位、LiFePO4、Li6PS5Cl toy。
测试任务分两类:
第一类是 ASE 结构弛豫,最多 50 步;
第二类是 300 K 短程 NVT MD smoke test,其中多数结构为 100 步,Li metal 额外跑到 1000 步。
后续如果需要自己实际做科研使用,可以在我的脚本上进行参数的更改,
3. 结构弛豫结果(R1):谁跑通,谁收敛
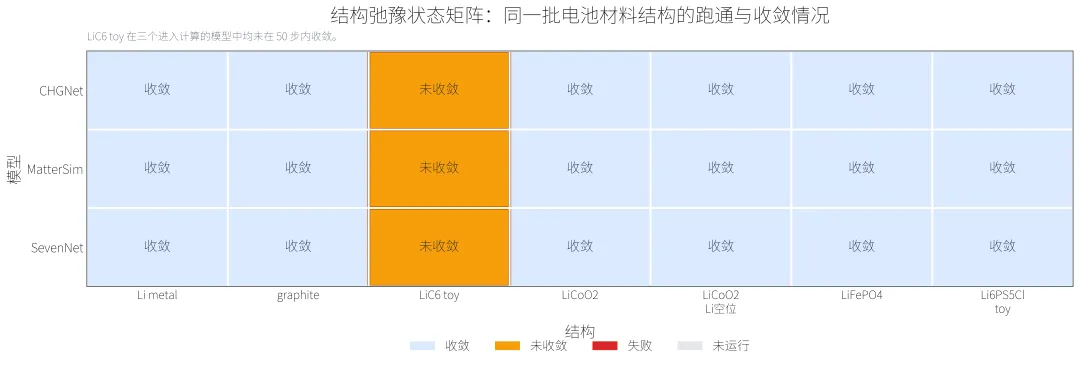
CHGNet、MatterSim、SevenNet 都完成了 7 个结构的弛豫尝试,并且各自有 6 个结构达到本次设置的收敛判据。
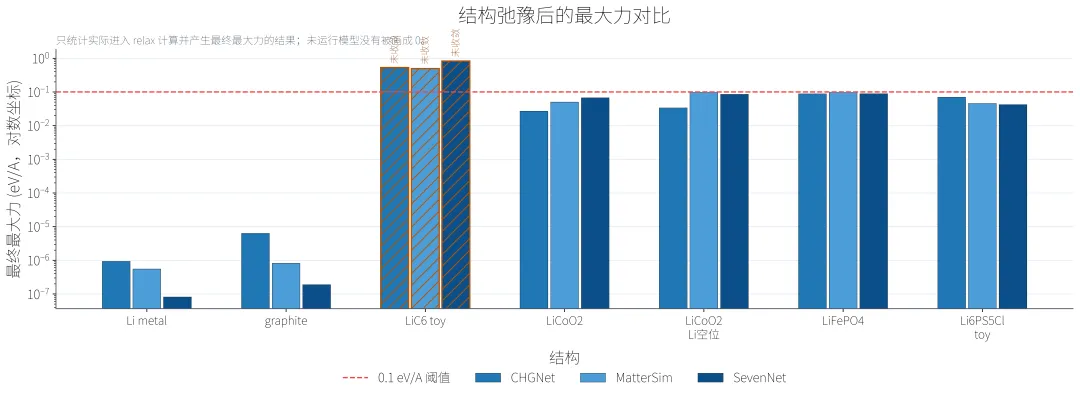
LiC6 toy 是这组结构里最明确的 relax 失败点。三种可运行模型都跑完了 50 步(还需要往下跑,用我的脚本,你自己动手试试),但都没有达到收敛:
CHGNet: 最终最大力 = 0.527 eV/A,50 步后是否收敛:否 MatterSim: 最终最大力 = 0.493 eV/A,50 步后是否收敛:否 SevenNet: 最终最大力 = 0.823 eV/A,50 步后是否收敛:否
这不等于模型完全不可用,但说明这个结构在当前设置下不能直接相信 relax 后结果。对实际科研来说,这类失败比平均指标很好看重要,因为它直接决定某个体系能不能进入后续计算。
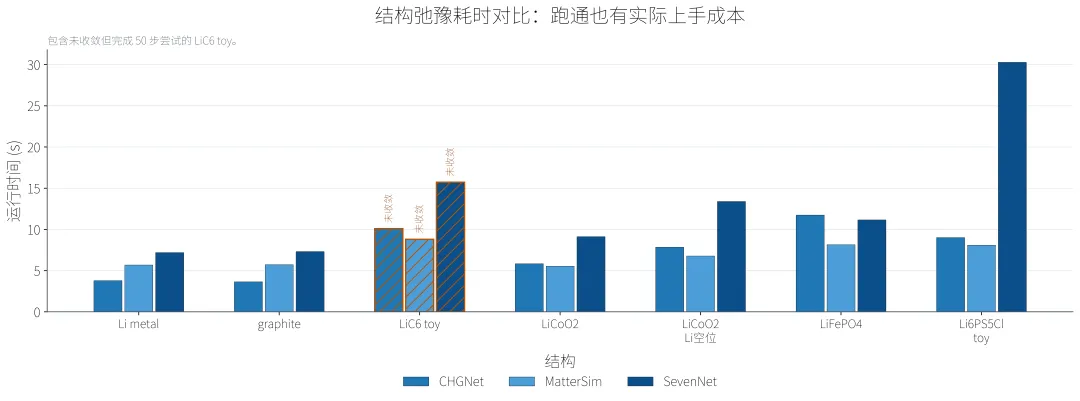
耗时图也说明了一个现实问题:跑通不只是精度问题,也有实际上手成本。这里所有测试都在我卡卡的写论文机器的 CPU PyTorch环境下完成,绝对时间不能外推到 GPU 生产环境,但同一机器、同一批结构下的对比能反映初次上手时的实际负担。
4. 短程 MD 结果(R2):能跑不等于能跑稳
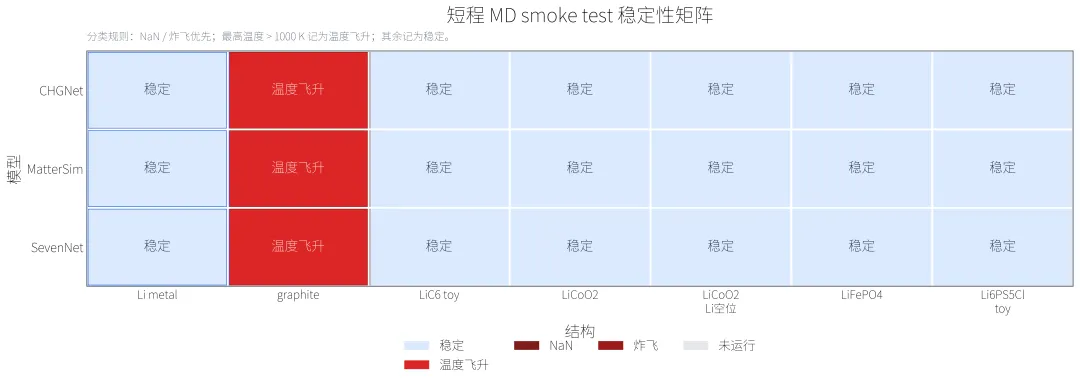
MD 短步长测试的核心结论很直接,没有 NaN、没有立刻炸飞,不代表就是稳定 300 K MD。graphite 100 步 MD 是本次最明显的失败点:
CHGNet: 100 步,最高温度 = 28163 K,能量漂移 = -0.2439 eV/atom MatterSim: 100 步,最高温度 = 21563 K,能量漂移 = -0.1551 eV/atom SevenNet: 100 步,最高温度 = 22882 K,能量漂移 = -0.3485 eV/atom
这些数值只说明本次 graphite toy 结构和当前 MD 设置下出现严重温度飞升,但这个结果竟供参考,可能是结构生成的不是很合理。
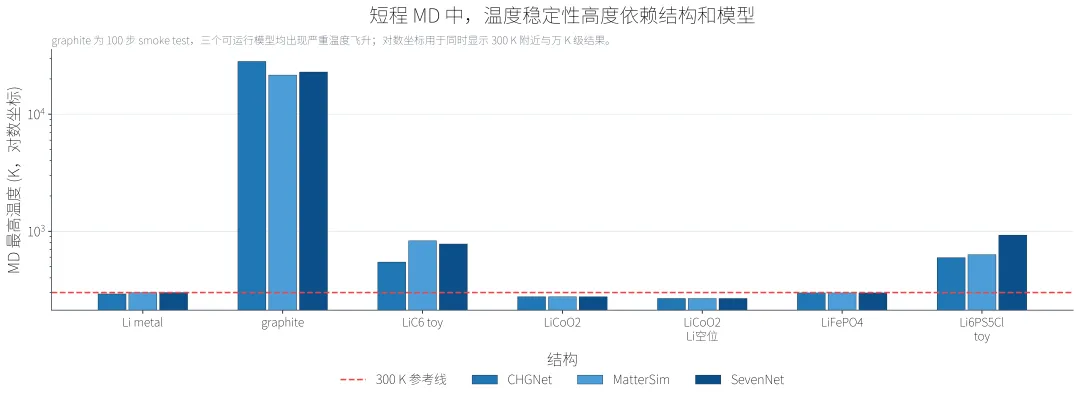
Li metal 是相对清楚的成功案例。它在三个可运行模型中都完成了 1000 步,并且无 NaN、未炸飞:
CHGNet: 1000 步,NaN:否,炸飞:否,最高温度 = 291.56 K MatterSim: 1000 步,NaN:否,炸飞:否,最高温度 = 299.12 K SevenNet: 1000 步,NaN:否,炸飞:否,最高温度 = 299.68 K
但 1000 步仍然只是短程检查,给大家一个初步的参考。
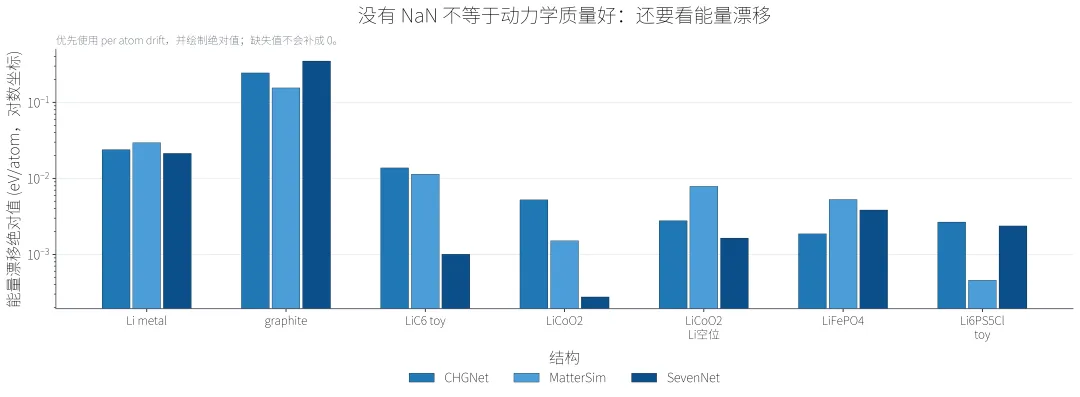
能量漂移图用于提醒各位,即使轨迹没有 NaN,也要看能量和温度是否合理。尤其是短程MD,如果温度已经大幅偏离目标温度,后续动力学性质不能直接拿来解释材料行为。
5. Discussion
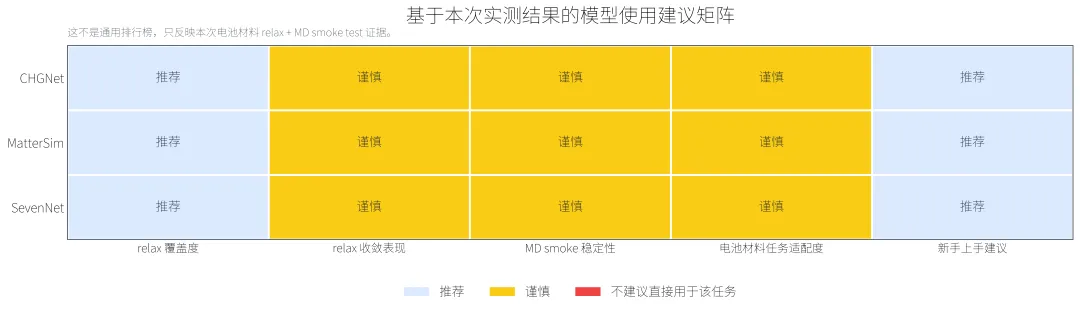
这张表并非是模型排行榜,而是基于本次电池材料的短程测试得到的实际使用建议。本轮测评只纳入CHGNet、MatterSim 和 SevenNet三个能够完成有效计算的模型,重点比较它们在小型电池材料结构上的安装可用性、结构弛豫表现、短程 MD稳定性和失败模式。
从本次结果看,CHGNet、MatterSim 和 SevenNet都可以作为电池材料结构快速筛选的候选工具。它们适合用于前期结构预筛、异常结构排查、候选构型初步弛豫,以及为后续 DFT或更高精度模拟提供初始结构。
实际科研使用时,更合理的策略不是简单选择宣传指标最高的模型,而是先在自己的体系内建立一个小型 benchmark。这个 benchmark 至少应包含已知稳定相、典型缺陷结构、界面结构、插层/脱嵌结构、低质量初始结构,以及少量可由 DFT 校准的参考样本。只有当模型在这些代表性结构上同时满足弛豫合理、力收敛正常、能量排序可信、短程 MD 不发生明显热失控,才适合作为后续高通量筛选或机理分析工具。
6. 写在最后
这次测评首先说明一件很朴素的事:大原子模型已经不是只能停留在论文里的东西了,普通材料人确实可以把它们拿出来跑一跑。
但也要说清楚,本次测评只是一个很小规模的测试。这里测试的 CHGNet、MatterSim、SevenNet,并不代表当前大原子模型的最强水平。现在这一类模型更新非常快,新模型在训练数据规模、元素覆盖、能量力精度、结构弛豫稳定性和动力学表现上,肯定还有更大的想象空间。因此,它更是一次真实上手记录,也就是在一台普通机器上,用一组小型电池材料结构,看看这些模型到底能不能装、能不能跑、哪里会出问题,手把手教一下刚入门这个方向的研究者,这个东西该怎么做。
后续我会把这类测试升级成面向真实材料场景的系统benchmark,不仅看能量和力的误差,也看安装成本、推理速度、显存占用、结构适配性、MD 稳定性、失败模式、下游筛选效率,以及模型在具体材料体系中的可解释边界。
对模型团队、材料课题组和企业研发来说,这类评估的意义不只是谁分高,而是把模型能力翻译成:
哪些体系适合用
哪些任务能加速
哪些结论可以写进报告
哪些地方还必须回到 DFT 或实验验证
至于更大规模、更高配置、更贴近真实科研和产业任务的测试,显然需要更完整的结构库、更稳定的计算资源和更明确的应用场景。后面如果有模型团队、材料课题组或企业研发部门希望把自己的模型或材料体系放进来做更深入的横向评估,欢迎后台联系我继续扩展成定制化benchmark、联合测评报告或面向应用场景的模型体检方案。
