 夜雨聆风
夜雨聆风各医疗器械相关单位:
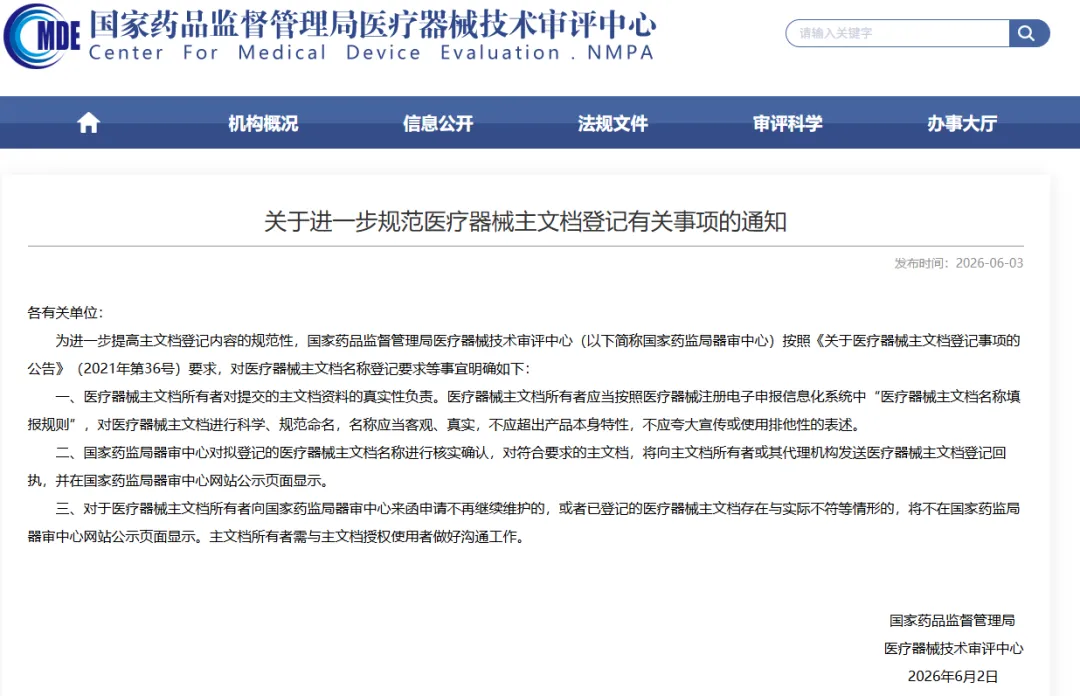
为进一步提高医疗器械主文档登记内容的规范性,2026年6月2日,国家药品监督管理局医疗器械技术审评中心(以下简称“国家药监局器审中心”)正式发布《关于进一步规范医疗器械主文档登记有关事项的通知》。
该通知自公布之日起施行,拟进行主文档登记的相关单位需密切关注以下核心变化。
核心要求:名称必须客观真实,严禁夸大宣传
本次通知重点在于规范主文档的名称登记。国家药监局器审中心明确要求:
责任主体更明确:医疗器械主文档所有者对提交的主文档资料的真实性负全部责任。
命名规则更严格:所有者必须按照电子申报系统中的“医疗器械主文档名称填报规则”进行命名。
禁止行为:名称必须科学、规范、客观、真实,不应超出产品本身特性,严禁夸大宣传或使用排他性表述(如“最佳”、“唯一”、“独家”等)。
监管升级:不合规将被“下架”处理
国家药监局器审中心将对拟登记的主文档名称进行核实确认。这意味着命名不再是简单的填写,而是进入了实质性的规范性审查阶段。
审核通过:对符合要求的主文档,将发送登记回执,并在中心网站公示页面显示。
不予显示:对于不再维护的申请,或者已登记的医疗器械主文档存在与实际不符等情形,将不再在公示页面显示。
在此提醒各主文档所有者,若因命名问题被撤销公示,不仅影响自身形象,还需自行与授权使用者做好沟通解释工作。
深度解读:为何要严管“命名”?
医疗器械主文档制度自2021年实施以来,极大方便了原材料等关键元器件的注册申报,避免了资料的重复提交。
然而,随着登记数量增加,部分主文档名称出现了夸大、误导或表述不规范的问题,扰乱了审评秩序。此次规范旨在回归制度初衷,确保主文档登记的严肃性和公信力。
特别提醒:主体责任的连带效应
虽然主文档登记是自愿行为,且登记回执仅证明“存档待查”,不代表技术审评通过,但这绝不意味着标准降低。
医疗器械终产品的安全性、有效性责任主体始终是医疗器械注册人(申请人) 。注册人需指导并协助主文档所有者建立合规文档。如果主文档名称不规范甚至虚假,将直接关联到后续的医疗器械注册审评进度。
2026年的这次规范通知,是国家药监局器审中心对医疗器械注册管理精细化、规范化的又一次迈进。
建议各主文档所有者和医疗器械注册人立即对照新规,自查已登记或待登记的主文档名称。如存在夸大、不实或排他性表述,应尽快通过“医疗器械主文档登记更新申请表”进行修正,以免影响后续关联审评及市场准入。
可凡得医疗科技有限公司是一家专业从事各类医疗器械管理体系咨询及提供产品研发辅助服务的公司,致力于为医疗器械企业提供最具价值的法规咨询与合规应对方案,帮助客户在中国市场获得可持续的竞争优势。公司拥有一支专业从事医疗器械研发、生产转换、工艺技术、产品申报注册、体系法规建设咨询的业务团队,团队成员全部拥有10年以上实际现场工作经验。
服务范围:
1.质量管理体系辅导培训: ISO13485、GB/T42601、QSR820、MDR;
2.产品注册咨询:国内产品注册、NMPA、FDA 510K、CE MDR、其他单一国注册;
3.医疗器械产品研发、技术转让、生产转换、工艺技术开发、技术咨询、技术交流、技术推广;
4.法规咨询培训:提供长期国内外医疗器械法规咨询;
5.合规咨询:产品注册策略咨询,经验引导咨询,专家支持咨询,设计开发全程咨询;
6.申报支持:注册支持(首次/变更/延续/界定),发补应急支持,临床评价资料支持;
7.咨询管理:合规咨询(策略/路径/方案),临床评价资料咨询;
8.第一类、第二类、第三类医疗器械生产、销售;
9.高新研发项目及费用管理;
10.翻译服务。
