 夜雨聆风
夜雨聆风
📁 准备输入文件运行需要两个核心文件:Ranked list 和 Gene set database。
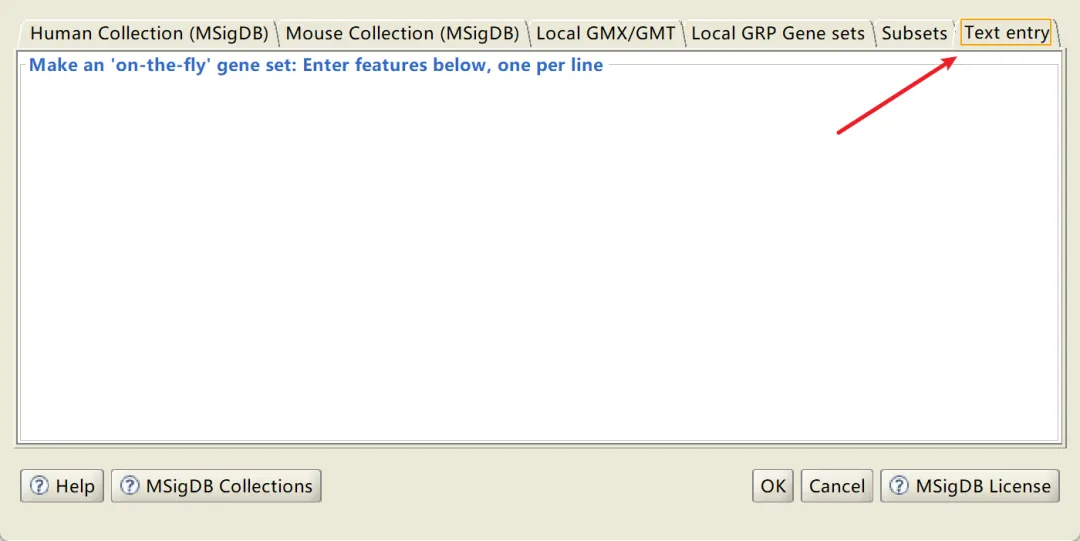
1️⃣ Gene set database (基因集文件)可以选用软件自带的数据库,也可以自定义。自定义格式很简单:一行一个基因名即可,如下演示所示
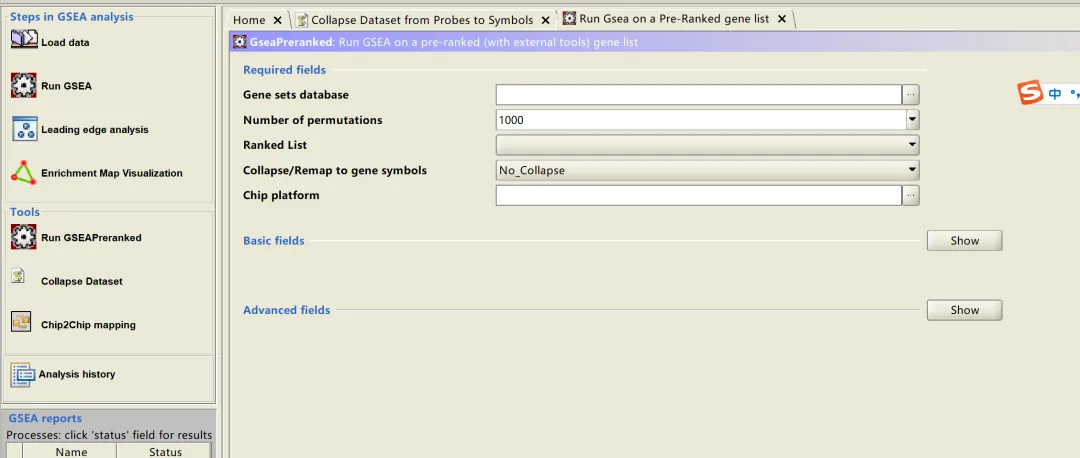
2️⃣ Ranked list (排序基因列表)这是重头戏!我们需要用Seurat的FindMarkers整理出.rnk文件。直接复制下方代码,替换你的目标细胞群即可运行:保存下来的rnk文件使用左上角的Load data导入
🏃 运行与查看结果检查无误后,点击左下角的 Run!
当左下角状态栏出现绿色的 Success 字样时,点击它就能直接查看丰富的富集结果报告啦!🎉
#out_dir定义下marker <- FindMarkers(seu,ident.1 = "your-cell-of-interest",min.pct = 0.05,logfc.threshold = 0.25,test.use = "wilcox",verbose = TRUE)selected_marker <- marker %>%dplyr::filter(p_val_adj < 0.05)selected_marker$gene <- rownames(selected_marker)ranked_export <- data.frame(gene = selected_marker$gene,avg_log2FC = selected_marker$avg_log2FC)ranked_export <- ranked_export[order(ranked_export$avg_log2FC, decreasing = TRUE), ]write.table(ranked_export,file = file.path(out_dir, "ranked_list.rnk"),sep = "\t",quote = FALSE,row.names = FALSE,col.names = TRUE)cat("Exported ranked list:", nrow(ranked_export), "genes → ranked_list.rnk\n")
🔬 GSEA结果怎么看?
1. 核心统计指标:NES & FDR
- NES (标准化富集分数)
:核心指标!绝对值越大,富集越强。 - NES > 0
:基因集在第一组(如上面定义的ident.1的细胞类型)中上调/富集。 - NES < 0
:基因集在第二组(如其他的细胞)中上调/富集。 - FDR q-value (错误发现率)
:控制假阳性的关键!q < 0.25 通常认为富集显著weibo.com+2。更严格可设为 < 0.05。
2. 显著性判断标准一个结果被认为显著富集,需同时满足:✅ |NES| > 1 (富集信号较强)✅ p值 < 0.05✅ FDR q值 < 0.25
错误发现率(FDR)的统计值(q值)表示该基因集的富集结果为假阳性发现的可能性(例如,若q = 0.25,则意味着发现的富集基因集中有25%可能是假阳性)。
NES的绝对值低于或接近1意味着无富集现象,这与大于0.25的相关q值所证实的结果一致。
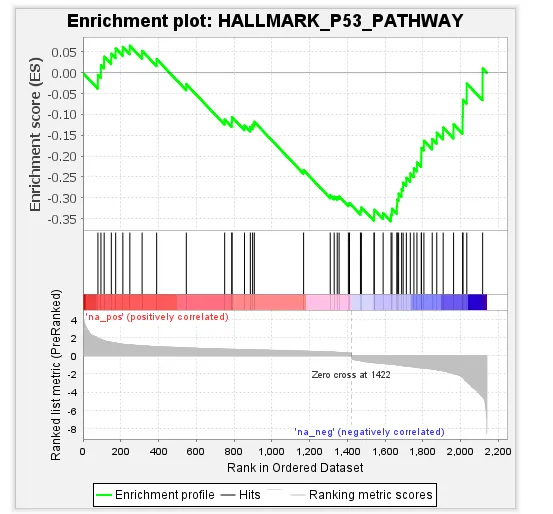
3. 可视化解读:条形码图GSEA的“条形码图”直观展示基因集的富集位置:
- 红色区
:基因在第一组中高表达(NES为正时富集侧)。 - 蓝色区
:基因在第二组中高表达。 - 绿色曲线峰值
:对应富集分数(ES),峰值位置越靠边,富集信号越强csdn.net。 - 黑色竖线
:代表基因集内的核心基因(Leading Edge),是驱动富集的关键。
4. 研究中的解读示例
“我们发现,Villani等人定义的DC3特征基因集(Gene Set)在DC3与DC2的比较中,NES = +1.8, FDR q = 0.12。这表明该DC3特征基因集在DC3中显著上调,且结果可靠(q < 0.25)。条形码图显示其核心基因(黑色竖线)紧密聚集在列表右侧(蓝色区),证实了DC3的特异性。”
💡 小贴士:GSEA关注的是基因集的整体协调变化,即使单个基因差异不显著,只要整体趋势一致,也可能得到显著富集结果。这正是它比传统差异基因富集分析更灵敏的原因!结果图如下
参考文献-Transcriptional and Functional Analysis of CD1c+ Human Dendritic Cells Identifies a CD163+Subset Priming CD8+CD103+TCells
