 夜雨聆风
夜雨聆风一、参考论文

使用的数据包括:
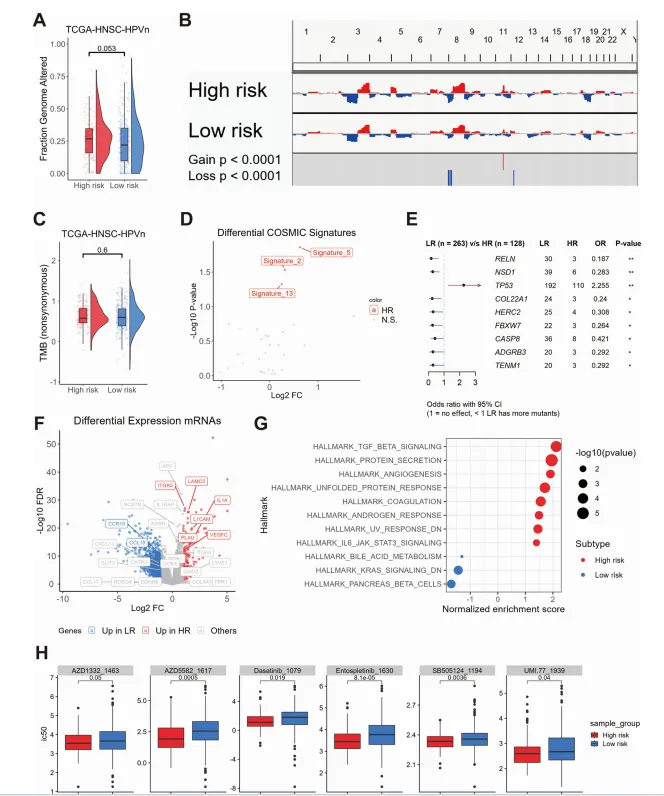
转录组数据:TCGA-HNSC队列的mRNA表达谱(395例HPV阴性样本)。
临床数据:TCGA-HNSC的总生存期(OS)、无进展间隔(PFI)、肿瘤分期、淋巴结转移、放疗信息等。
基因组数据:体细胞突变(含COSMIC突变特征)、拷贝数变异(CNV)、肿瘤突变负荷(TMB)、基因组改变分数。
单细胞RNA-seq数据:来自GEO的三个公共数据集(GSE181919、GSE182227、GSE234933),共34例HPV阴性样本的115,479个细胞。
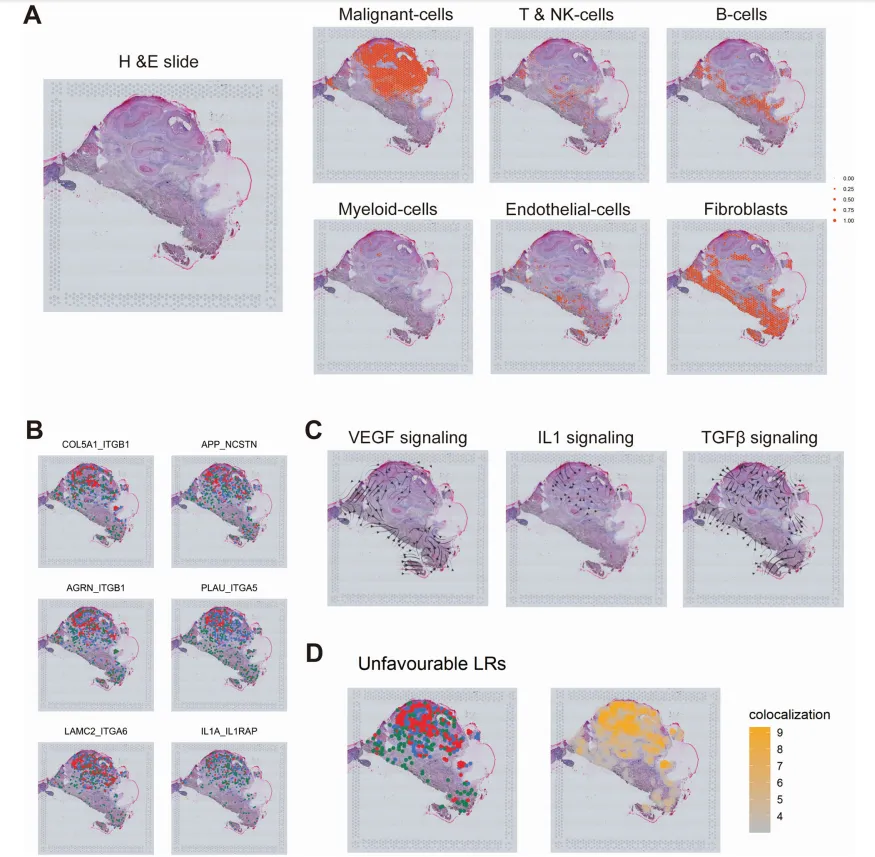
空间转录组数据:GEO数据集GSM5494476(GSE181300),用于配体-受体空间共定位分析。
组织病理图像:TCGA-HNSC的348例HPV阴性病例的360张HE染色全切片图像(WSIs)。

二、复现及创新
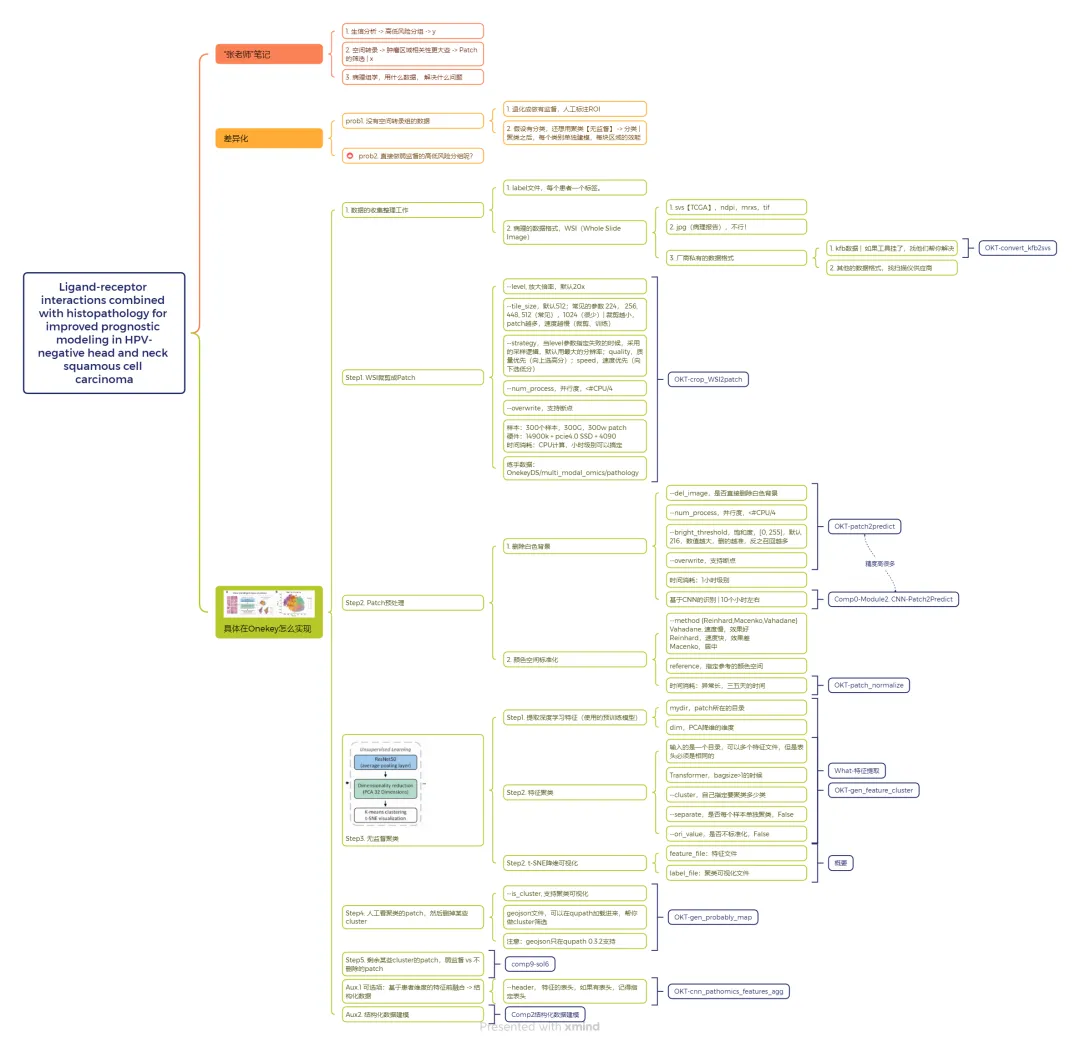
OneKey平台复现论文的核心步骤
1. 数据准备与预处理
病理图像格式要求:
必须是全切片图像(WSI),常见格式为.svs、.ndpi、.mrxs。
不支持单张截图(如.jpg、.png),除非是私有格式如.kfb(江丰生物),可通过工具转换为.svs。
数据收集:
使用公开数据集(如TCGA-HNSC)或私有数据。
需要准备患者标签(label)
2. WSI裁剪为Patch
工具:WsiToPatch
参数说明:
level:放大倍率,TCGA数据一般为20倍(对应 level=0 或 level=1)。
patch_size:裁剪大小,常用512×512像素。
strategy:若指定倍率失败,默认使用最高分辨率。
输出:每个WSI被裁剪为数千个patch,用于后续分析。
3. Patch预处理
删除白色背景:
工具:PatchToPredict
参数:saturation_threshold 默认216,数值越大删除越严格。
颜色空间标准化:
工具:StainNormalize
常用方法:Macenko(效果好但慢)、Vahadane(居中)、Reinhard(快但效果差)。
需要指定参考图像(reference)进行颜色归一化。
4. 无监督聚类(核心差异化步骤)
目的:识别具有生物学意义的病理区域(如肿瘤区、间质区、浸润前沿等)。
工具:
FeatExtract:提取深度学习特征(使用预训练模型如ResNet50)。
FeatureCluster:对特征进行K-means聚类,指定聚类数(如6类)。
DimReduction + TSNE:可视化聚类结果。
输出:每个patch的聚类标签及距离聚类中心的距离。
5. 聚类结果筛选(Silicon病理区域选择)
人工干预:
根据聚类结果和病理学知识,筛选出有意义的聚类(如肿瘤区、浸润前沿)。
删除无关区域(如肌肉、脂肪、背景)。
工具辅助:
使用QuPath 或 PatchVisualizer 查看每个聚类的代表性patch。
可通过cluster_to_wsi 工具将聚类结果映射回原图,辅助判断。
6. 弱监督学习建模
目的:基于筛选后的patch,预测患者风险(高/低风险)。
工具:WeaklySupervised
模型:支持多种CNN架构(ResNet18/50、Inception_v3)。
训练策略:
使用患者级别标签(来自生信分析的风险分组)。
采用多实例学习(MIL)或特征聚合(如FeatAggregate)进行训练。
评估:对比筛选前后模型的AUC、准确率、混淆矩阵等。
7. 结果可视化与解释
Grad-CAM:生成热图,显示模型关注的病理区域(如血管浸润、间质浸润)。
聚类映射:将聚类结果可视化到WSI上,便于病理医生验证。
工具:WSIHeatmap、QuPath、Matplotlib。

三、视频讲解
第一节
第二节

