 夜雨聆风
夜雨聆风1 细胞不再只是“固定类别”:单细胞时代,免疫学需要一朵“细胞云”
❝文章:The cell cloud: Adopting systems biology concepts in the era of single-cell immunology
链接:https://doi.org/10.1371/journal.pbio.3003853
过去一百多年里,生物学家习惯把细胞看作一个个清晰的“类型”:肝细胞、T细胞、B细胞、中性粒细胞……在免疫学中,这种分类方式尤其深入人心。研究人员常常依靠显微镜形态、流式细胞术中的表面标志物,或特定转录程序,把细胞划入不同类别。与此同时,同一种细胞在不同环境、代谢状态、细胞周期或昼夜节律下出现的短暂变化,则被称为“细胞状态”。这种“细胞类型加细胞状态”的框架曾极大推动了免疫学发展。
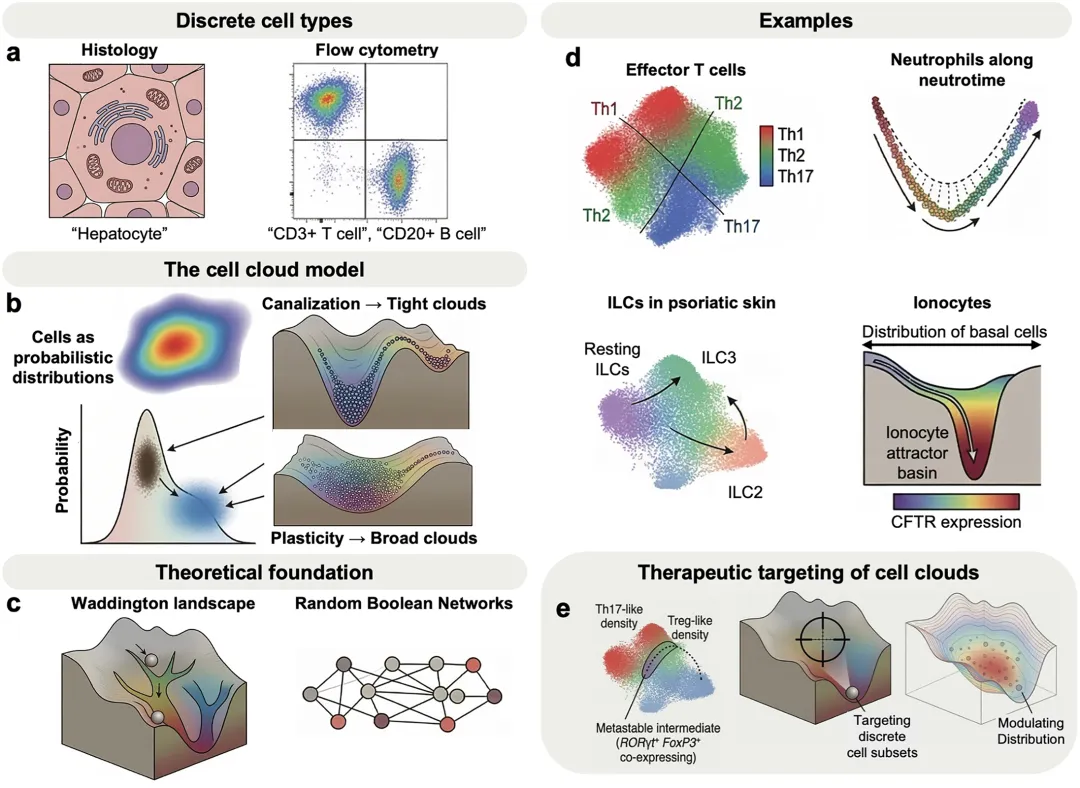
然而,单细胞RNA测序等高通量技术正在改变这一经典图景。当研究人员能够同时检测数百万个细胞、数千个基因特征时,他们发现很多细胞并不像教科书中那样整齐地分成几类,而是分布在一个连续的转录空间中。有些区域细胞密度很高,对应传统意义上的细胞类型;有些区域细胞较少,可能代表过渡状态或不稳定中间状态。基于这一观察,发表在 PLOS Biology 的这篇观点文章提出了“细胞云”模型:细胞类型并不是绝对孤立的格子,而是细胞在基因表达空间中以一定概率占据的一片“云”。
作者指出,“细胞云”并不是完全推翻细胞类型概念,而是让它变得更符合真实生物学。其理论基础可以追溯到系统生物学中的Waddington表观遗传景观和Kauffman随机布尔网络。通俗地说,细胞分化就像小球沿山谷滚动,不同山谷代表不同稳定命运。如果山谷又深又窄,细胞状态会很稳定,形成紧密的细胞云;如果山谷较浅,细胞就更容易摆动和转换,形成更宽、更模糊的细胞云。传统细胞类型可以理解为这些连续景观中的高密度区域,而不是完全割裂的岛屿。
文章以多个免疫学例子说明这种连续性并非测序噪音。经典的辅助性T细胞亚群,如TH1、TH2和TH17,在实际数据中常呈连续分布,强行划分边界往往带有人为性。中性粒细胞也可沿着一个被称为“neutrotime”的时间轨迹连续变化,而不是天然分成固定小群。在银屑病皮肤中,先天淋巴细胞并不严格分为ILC1、ILC2和ILC3,而是形成连续云状结构,其中ILC3样细胞可由ILC2样细胞或静息ILC转变而来。这些结果说明,细胞身份的连续变化本身就是重要生物学信号。
当然,“细胞云”模型也承认真正离散的稀有细胞类型仍然存在。例如肺离子细胞就是通过单细胞测序发现的罕见细胞类型,它高表达CFTR等离子运输相关基因,并与囊性纤维化发病相关。它虽然处于与基底细胞相连的发育轨迹中,但占据一个较深、较窄的稳定区域,因此仍可被视为清晰细胞类型。
作者还提醒,研究者必须区分真实生物学变异和技术噪音。转录爆发会让同类细胞在不同采样时刻显示不同RNA水平,而单细胞测序中的dropout、测序深度不足等技术问题也会人为拉宽“细胞云”。因此,大规模细胞图谱适合描绘整体景观,但若要解析某一类细胞内部的细微梯度,则需要更深的测序覆盖。
这一模型对免疫治疗具有重要启发。过去药物常针对几个表面标志物定义的细胞群,但这往往只命中了“云”的中心区域,忽略了边缘状态和过渡细胞。比如同时表达RORγt和FOXP3的细胞可能代表TH17与调节性T细胞之间的不稳定中间态,这类细胞或许正是疾病干预的新靶点。总体而言,“细胞云”提醒我们,免疫细胞身份不是静态标签,而是由基因调控网络、环境信号和随机波动共同塑造的动态概率分布。单细胞时代的免疫学,或许需要从“给细胞贴标签”,走向理解细胞身份如何生成、维持与转变。
2 让“旧数据”长出空间坐标:HistoMap从普通RNA测序中重建肿瘤单细胞地图
❝文章:HistoMap: Reconstructing Spatially Resolved Single-Cell Profiles from Bulk RNA-Seq to Decipher the Immune-Excluded Microenvironment in Colon Cancer
链接:https://doi.org/10.3390/ijms27125259
代码:https://github.com/stat-hj/HistoMap.git
在过去二十多年里,bulk RNA-seq积累了海量公共数据,例如TCGA、ICGC等大型数据库中保存了成千上万份肿瘤转录组资料。但这类数据有一个天然缺陷:它测到的是一块组织中所有细胞的平均表达信号,就像把一座城市的所有声音混在一起,只能听到总体噪声,却难以分辨每个人在哪里、说了什么。相比之下,单细胞测序和空间转录组能揭示细胞异质性和组织结构,但成本高、样本量有限、实验周期长。如何把已有的bulk RNA-seq重新“激活”,推断出其中隐藏的单细胞组成和空间分布,成为当前计算生物学的重要问题。
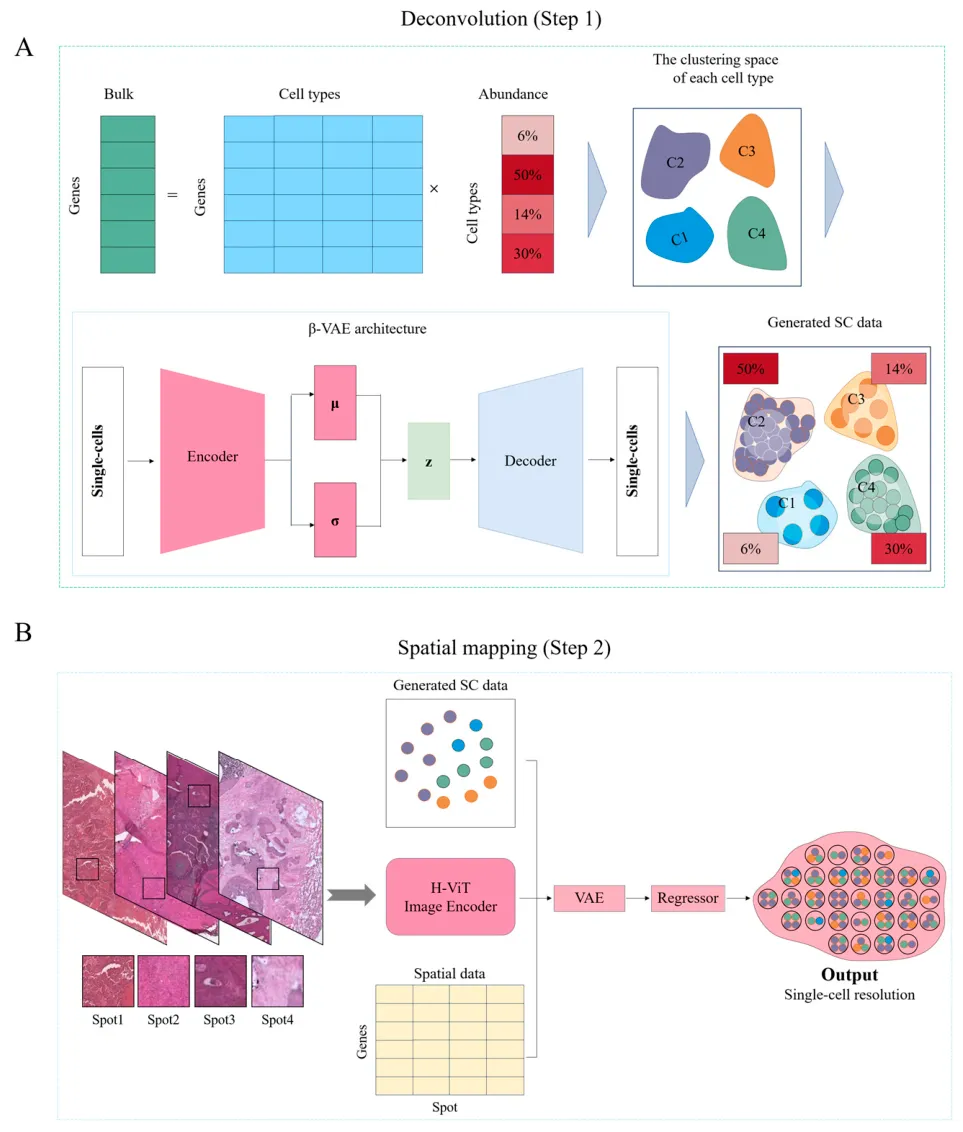
这项发表在 International Journal of Molecular Sciences 的研究提出了一个深度学习框架HistoMap,试图打通bulk RNA-seq、单细胞RNA测序、空间转录组和病理切片图像之间的壁垒。简单来说,HistoMap希望从普通bulk转录组数据出发,先“拆解”出高保真的伪单细胞表达谱,再结合H&E染色病理图像和空间转录组参考,把这些细胞放回组织中的可能位置,最终生成一张具有空间分辨率的单细胞表达地图。
HistoMap采用两阶段流程。第一阶段使用β-变分自编码器,也就是β-VAE模型,以单细胞RNA测序数据作为参考,从bulk RNA-seq中重建细胞级表达谱。传统CIBERSORT、MuSiC等工具通常只能估计“某类细胞有多少”,而HistoMap更进一步,尝试生成接近真实单细胞状态的表达数据。第二阶段则使用组织学视觉Transformer,也就是H-ViT,从H&E染色图像中提取组织形态特征,再结合空间转录组参考和组织拓扑约束,预测每个重建细胞最可能出现的空间坐标。换句话说,它不仅回答“有哪些细胞”,还回答“这些细胞可能在哪里”。
研究人员在脑、乳腺、肝、肺、肠、扁桃体、前列腺和皮肤等8类人类组织数据上评估模型性能。结果显示,在单细胞重建任务中,HistoMap相比GAN和条件GAN具有更高的表达相关性和更低误差,并在十折交叉验证中表现稳定。在空间映射任务中,HistoMap与Tangram、scSpace、SPOTlight和Bulk2Space等方法相比也表现最佳,外部验证中Pearson相关系数达到0.800,十折交叉验证中平均PCC可达0.832,说明其在不同组织和样本中具有较好的泛化能力。
研究还通过消融实验强调了多模态整合的重要性。仅使用基因表达信息或仅使用病理图像,模型性能都会下降;完整HistoMap表现最佳。值得注意的是,仅使用图像的OnlyIma模型表现甚至优于仅使用基因的OnlyGene模型,提示H&E病理图像中蕴含着丰富的空间结构信息,可为细胞定位提供重要线索。
在结直肠癌案例研究中,HistoMap对14个病例进行了单细胞空间重建,生成的“伪单细胞”在UMAP空间和细胞标志基因表达上与参考单细胞数据高度一致。更重要的是,空间图谱揭示了一个由SPP1阳性巨噬细胞和肌成纤维细胞共同构成的促纤维化屏障。这些SPP1+巨噬细胞富集在肿瘤与间质交界的侵袭前沿,像“空间枢纽”一样与成纤维细胞、髓系细胞紧密相邻,形成围绕肿瘤巢的连续隔离带。
这一屏障具有明确的免疫学意义。尽管样本中存在CD4+ T细胞、CD8+ T细胞和B细胞,但它们大多被限制在远端间质或免疫细胞聚集区,难以进入肿瘤核心。定量空间分析显示,Macro_SPP1主要分布在距离肿瘤边界约50至100像素的区域,而在这些细胞和成纤维细胞占据的区域内,细胞毒性CD8+ T细胞密度迅速下降。这提示SPP1+巨噬细胞—成纤维细胞轴可能是造成结直肠癌“免疫排斥型”微环境的重要原因,也解释了部分患者对免疫检查点抑制剂反应不佳。
HistoMap的意义不仅在于提出了一个新算法,更在于为海量历史bulk RNA-seq数据赋予了新的空间解释能力。它有望帮助研究者从已有数据中重建组织结构、解析细胞互作,并为肿瘤免疫治疗提供新的靶点线索,例如联合阻断SPP1信号、TGF-β驱动的基质重塑或抗血管生成治疗,从而打破免疫细胞进入肿瘤的物理和化学屏障。
3 单细胞“自动识别”再添新工具:scGMB让细胞分类更快、更准、更懂细胞关系
❝文章:scGMB: A scRNA-seq Cell Classification Method Combining GCN and Mamba
链接:https://doi.org/10.1049/syb2.70079
代码:https://doi.org/10.5281/zenodo.3357167
单细胞RNA测序正在帮助科学家以前所未有的分辨率认识组织和疾病,但海量细胞数据带来了一个现实难题:如何准确判断每个细胞属于哪一种类型?过去常用的无监督聚类方法需要研究人员根据标记基因手动注释,不仅耗时,而且容易出错。尤其是在复杂样本中,一个聚类里可能混入多种细胞状态或相近细胞类型,如果简单认为“同一簇就是同一类细胞”,就可能造成误判。因此,开发更稳定、更高效的自动细胞分类方法,已成为单细胞数据分析中的重要需求。
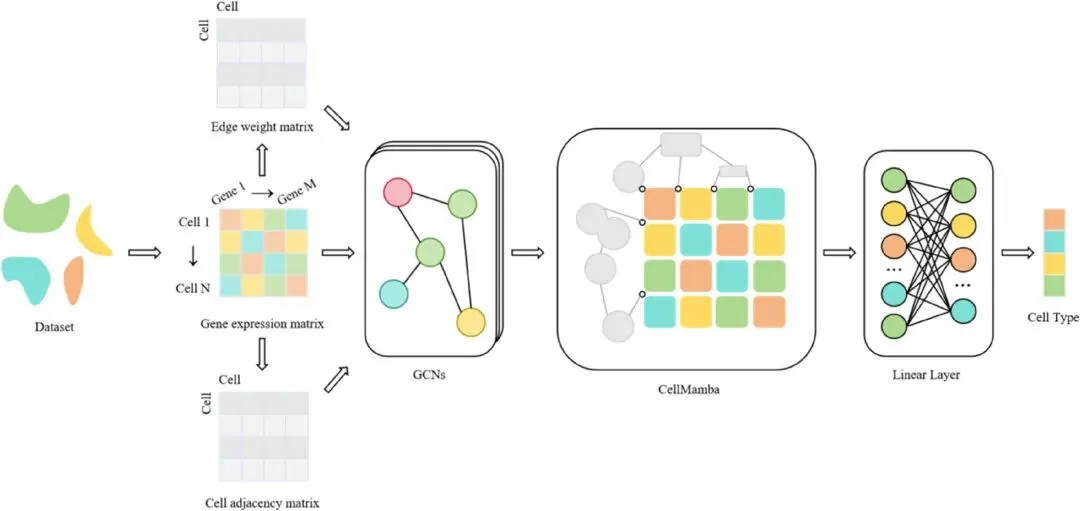
近日,发表于 IET Systems Biology 的一项研究提出了一种新的单细胞RNA测序细胞分类方法scGMB。该方法将图卷积网络GCN与近年来受到关注的Mamba模型结合起来,既能捕捉细胞之间的关系网络,又能高效处理单细胞数据中常见的高维、稀疏和长序列特征。简单来说,scGMB不仅看每个细胞自身的基因表达,还会参考与它相似或相关的邻近细胞,从而获得更完整的细胞身份判断依据。
scGMB的核心思路分为两部分。首先,研究人员根据基因表达矩阵构建“细胞图”,把每个细胞看作图中的一个节点,如果两个细胞共享非零表达基因,就在它们之间建立连接,并用共享基因表达信息作为边权重。随后,GCN模块会聚合邻近细胞的信息,让每个细胞的特征表示不仅包含自身表达模式,也包含周围相关细胞的上下文信息。这种设计比单纯依赖欧氏距离或余弦相似度更直接地利用了真实基因表达信号,也提高了模型的生物学可解释性。
第二部分是CellMamba模块。传统Transformer或BERT类模型虽然强大,但处理长序列时计算成本较高。Mamba基于选择性状态空间模型,能够以更低计算负担处理长序列数据。研究团队针对单细胞表达矩阵的二维结构进行了改造,使其更适合“细胞 × 基因”这类高维稀疏数据。CellMamba通过线性投影、一维卷积和状态空间模型,进一步提取基因表达中的局部模式和复杂动态特征,最后经过归一化、池化和分类器输出每个细胞的类型预测结果。
为了验证性能,研究人员在五个经典单细胞基准数据集上测试了scGMB,包括人外周血单个核细胞Zheng68K、流式分选PBMC数据Zhengsorted、人胰腺BaronHuman、小鼠胰腺BaronMouse和小鼠初级视觉皮层AMB。结果显示,scGMB在这五个数据集上的分类准确率分别达到72.2%、91.1%、98.9%、99.2%和99.5%。与SVM、LDA、SingleR、CHETAH、ACTINN以及scGraph、sigGCN、HNNVAT、scBIGNN等方法相比,scGMB在多数数据集上取得最佳表现,尤其在Zhengsorted、BaronHuman、BaronMouse和AMB数据集中优势明显。
研究还专门评估了GCN模块的贡献。去掉GCN后,模型在五个数据集上的准确率均有所下降,例如Zhengsorted从91.1%降至89.7%,BaronMouse从99.2%降至98.4%。这说明细胞间拓扑关系确实有助于提升分类效果。进一步的混淆矩阵、t-SNE可视化和ROC曲线分析也显示,scGMB在大多数细胞类型上误分类较少,不同细胞类型在低维空间中分离清晰,多数类别AUC接近1,说明模型具有较好的稳定性和区分能力。
scGMB的意义在于把“细胞关系网络”和“高效序列建模”结合到单细胞分类任务中。它不仅提高了自动注释的准确率,也为处理大规模单细胞数据提供了更高效的计算方案。作者表示,未来将把scGMB扩展到空间转录组和多组学数据分析中,进一步服务于细胞类型发现、疾病机制解析和生物医学研究。
4 综述:当“AI大模型”遇上单细胞:Transformer正在重塑单细胞组学分析
❝文章:Transformers for single-cell RNA sequencing: a survey
链接:https://doi.org/10.1093/bib/bbag332
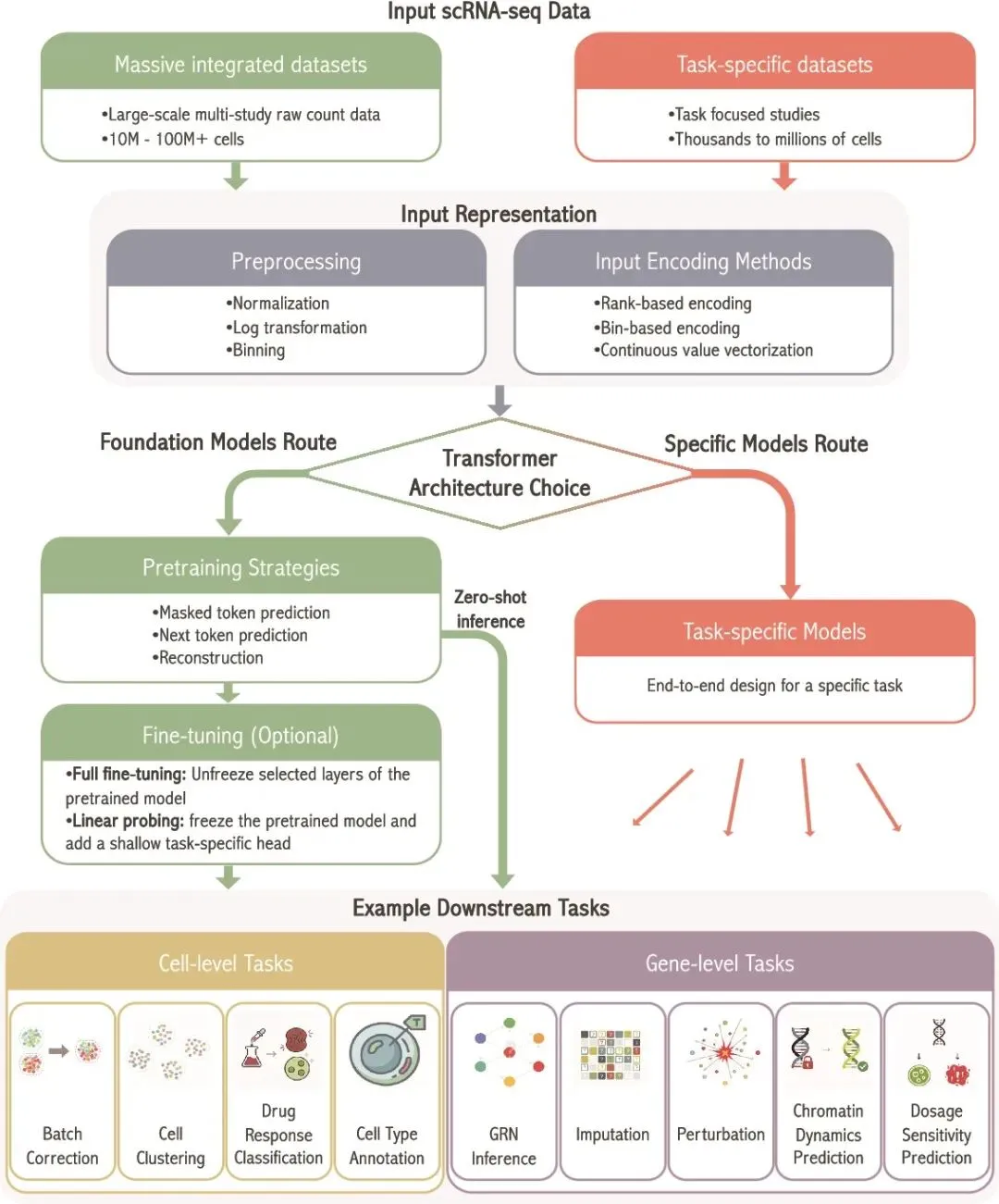
随着单细胞RNA测序技术快速发展,科学家已经能够在单个细胞水平读取成千上万个基因的表达信息,从而解析疾病进展、肿瘤微环境、发育轨迹和细胞异质性。然而,单细胞数据也天然“难处理”:基因数量动辄上万,细胞数量可达百万级,表达矩阵高度稀疏,还容易受到dropout和批次效应影响。传统神经网络和常规机器学习方法在面对这类高维、稀疏、复杂依赖的数据时,往往难以充分捕捉基因之间、细胞之间的全局关系。近日发表于 Briefings in Bioinformatics 的综述文章系统梳理了Transformer在单细胞RNA测序分析中的应用,指出这一类模型正在成为单细胞计算领域的重要推动力。
Transformer最初诞生于自然语言处理领域,其核心优势是自注意力机制。通俗来说,它可以让模型在分析一个“词”时,同时关注句子中其他所有“词”的关系。迁移到单细胞领域后,研究人员可以把基因看作“词”,把一个细胞的基因表达谱看作“句子”,从而学习基因之间复杂的依赖关系。相比传统模型,Transformer更擅长捕捉长距离依赖,也更适合通过大规模预训练学习通用表示,再迁移到细胞类型注释、批次校正、扰动预测、基因调控网络推断等下游任务中。
文章将现有单细胞Transformer模型分为两大类:一类是面向特定任务的模型,另一类是可适配多种任务的基础模型。基础模型代表包括Geneformer、scGPT和scFoundation。Geneformer使用约2990万个单细胞转录组进行预训练,通过基因表达排序构建输入序列,能够用于细胞类型注释、基因剂量敏感性预测、染色质动态预测和网络动态分析。scGPT基于约3300万个细胞进行生成式预训练,可支持细胞类型注释、批次整合、多组学整合、扰动响应预测和基因调控网络推断。scFoundation规模更大,使用超过5000万个单细胞数据,包含约1亿参数,并引入read-depth-aware策略,以适应单细胞测序深度差异,可用于细胞注释、读深增强、药物反应预测和扰动预测等任务。
除了基础模型,综述还介绍了多种任务特异性Transformer。例如scBERT较早将BERT式预训练用于单细胞细胞类型注释,并利用Performer降低长基因序列带来的计算压力。TOSICA则把基因映射到通路层面,使注意力权重可以追溯到具体生物通路,增强了可解释性。T-GEM可从基因表达中发现癌症相关标志基因和通路枢纽。CIForm通过类似图像分块的策略处理高变基因,提高大规模细胞注释效率。DeepMAPS进一步将Transformer与异质图结合,用于单细胞多组学中的生物网络推断。
文章强调,Transformer在单细胞领域的价值不仅是“更准”,还在于其可迁移性和可解释性。通过预训练,模型可以从海量公共单细胞数据中学习通用的细胞和基因表示,再用少量标注数据适配新任务。注意力机制则为解析关键基因、通路、调控网络和细胞状态特异性关系提供了可能。已有研究显示,在细胞类型注释和聚类任务中,部分Transformer模型优于传统方法;在基因层面,它们也逐渐用于扰动预测、基因调控网络推断和疾病相关基因优先级排序。
不过,挑战同样明显。标准Transformer的计算复杂度会随着输入长度平方增长,而单细胞数据常包含上万基因,因此预训练大模型需要大量GPU资源。例如Geneformer需使用12张V100 GPU训练3天,部分更大模型训练时间可达数十天。为降低成本,研究者正在使用Performer、FlashAttention、混合精度训练、DeepSpeed、低秩适配等技术提高效率。与此同时,许多模型仍直接借鉴自然语言处理架构,并未完全针对单细胞数据的稀疏性、高维性和批次效应定制,这也是未来的重要改进方向。
综述还指出,Transformer基础模型正在从scRNA-seq扩展到单细胞ATAC-seq、空间转录组和DNA甲基化等多组学领域。例如EpiAgent面向单细胞表观基因组,scGPT-spatial面向空间转录组,MethylGPT和CpGPT则探索甲基化数据建模。未来,真正强大的单细胞AI模型可能不再只读取基因表达,而是同时整合转录、染色质开放性、空间位置、甲基化和文本知识,构建更全面的细胞身份与调控机制图谱。总体来看,Transformer正在推动单细胞分析从“数据整理和注释”迈向“机制理解和预测生物学”的新阶段。
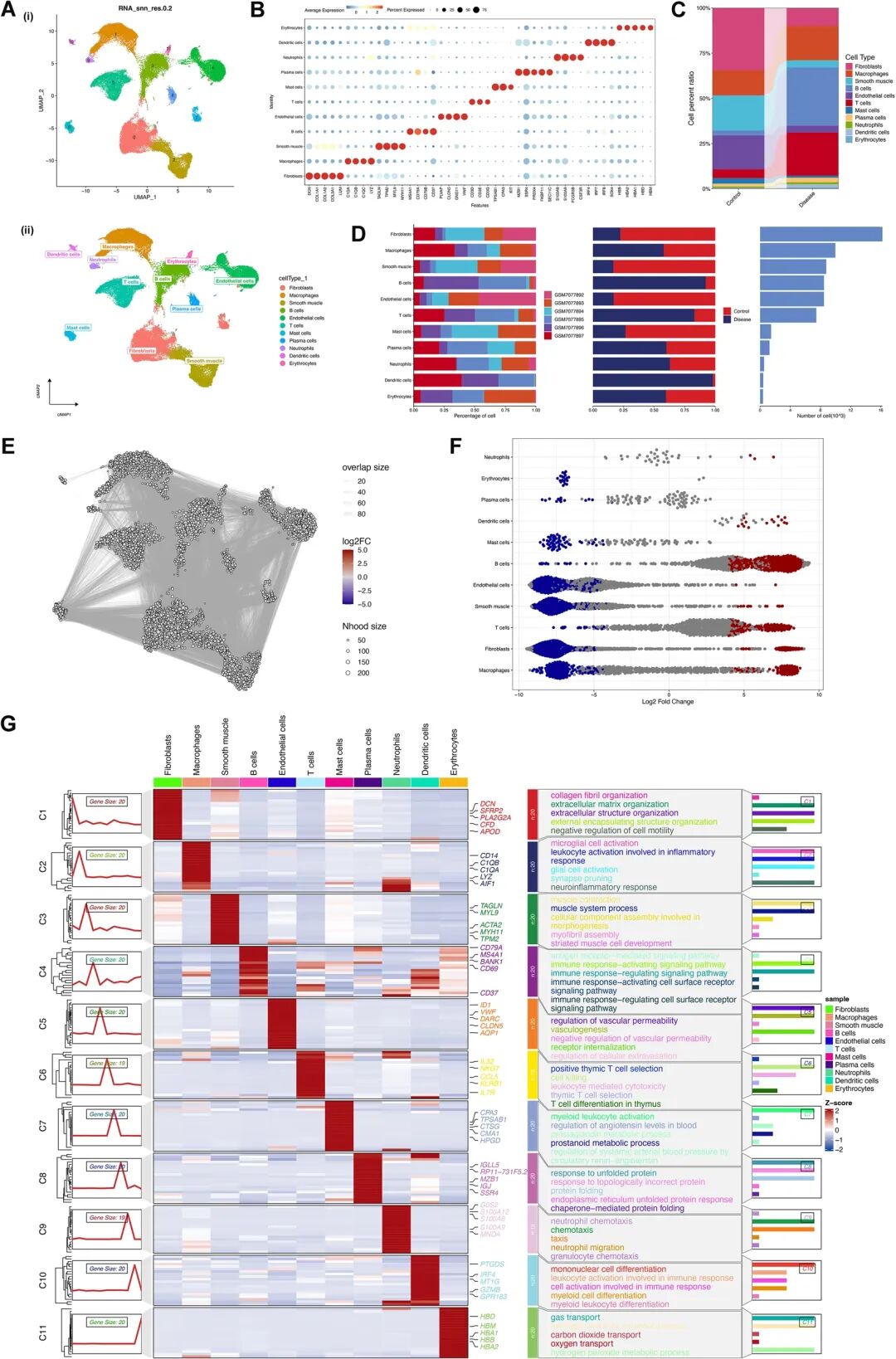
5 单细胞测序锁定“危险B细胞”:LUM⁺ B细胞或推动腹主动脉瘤恶化
❝文章:Single-cell sequencing combined with transcriptome analysis unravels LUM+ B cells as key drivers in abdominal aortic aneurysm
链接:https://doi.org/10.3389/fimmu.2026.1836487
腹主动脉瘤是一种隐匿而危险的血管疾病,表现为腹主动脉局部不可逆扩张。一旦破裂,死亡率可高达67%至94%。目前,对于较大或高风险动脉瘤,临床主要依赖开放手术或腔内修复,但这些治疗方式创伤较大,也可能带来内漏、感染、血栓等并发症。因此,寻找能够解释疾病进展的新机制,并开发非手术干预靶点,是腹主动脉瘤研究中的重要方向。
近日发表于 Frontiers in Immunology 的一项研究,将单细胞RNA测序、bulk转录组分析、机器学习筛选、细胞通讯分析和体外功能实验结合起来,发现一种表达LUM的B细胞亚群可能是推动腹主动脉瘤进展的重要力量。研究团队分析了GEO数据库中的AAA单细胞数据和转录组数据,共获得63,335个高质量细胞,并将其分为成纤维细胞、巨噬细胞、血管平滑肌细胞、B细胞、内皮细胞、T细胞、肥大细胞、浆细胞、中性粒细胞、树突状细胞和红细胞等主要细胞群。结果显示,与正常腹主动脉相比,AAA组织中巨噬细胞、B细胞、浆细胞、中性粒细胞和树突状细胞明显增多,而成纤维细胞、内皮细胞、平滑肌细胞和肥大细胞更多见于正常组织。
研究最重要的发现集中在B细胞上。通过MiloR差异丰度分析,团队发现B细胞不仅数量发生变化,其局部状态和分布也在AAA中显著改变。CytoTRACE2和Monocle2轨迹分析进一步显示,AAA中的B细胞更偏向分化中晚期状态,提示它们可能并非普通静息B细胞,而是处于活化、效应化的疾病相关状态。这些B细胞相关基因富集于抗原受体介导信号、免疫激活信号和免疫调节信号等通路,说明B细胞可能在AAA慢性炎症环境中持续被激活。
细胞通讯分析进一步揭示了B细胞的“枢纽”作用。AAA组织中的细胞间通讯明显增强,B细胞与成纤维细胞、内皮细胞、树突状细胞和血管平滑肌细胞之间的交流尤其频繁。研究发现,B细胞与这些细胞之间多个配体—受体通路被激活,其中TNF–TNFRSF1A和PPIA–BSG在多组细胞互作中持续上调。这意味着B细胞可能通过炎症因子和细胞间信号,放大局部血管壁炎症,并影响血管结构重塑。
为了寻找关键分子,研究人员在B细胞中筛选差异表达基因,并结合LASSO和SVM机器学习方法,最终锁定两个核心基因:CD79A和LUM。CD79A是B细胞受体复合体的重要组成部分,提示B细胞受体信号可能参与AAA免疫激活。LUM则更引人关注,它编码Lumican,是一种细胞外基质相关蛋白,过去多与胶原纤维组织、基质重塑和炎症过程有关。研究发现,LUM在AAA组织中无论mRNA还是蛋白水平均显著升高,免疫组化显示其在浸润免疫细胞中富集。进一步分离外周血CD79A阳性B细胞后,研究者发现AAA患者B细胞中的LUM表达显著高于正常对照。
更关键的是,LUM与疾病严重程度相关。研究将AAA患者按最大瘤体直径分为小、中、大直径组,结果显示B细胞中的LUM蛋白水平随动脉瘤直径增大而逐步升高,并与最大AAA直径呈显著正相关。血浆中的LUM水平也在AAA患者中升高。这提示LUM不仅可能参与疾病机制,也可能成为反映AAA严重程度的潜在生物标志物。
研究团队进一步通过体外实验验证LUM的功能。他们分离人外周血B细胞,利用慢病毒敲低或过表达LUM,再与原代人主动脉血管平滑肌细胞进行Transwell共培养。结果显示,普通B细胞可促进平滑肌细胞从“收缩型”转向“合成型”,表现为合成型标志物OPN升高,而收缩型标志物αSMA、CNN和SM22α下降。这种表型转换是AAA发生发展的关键事件,因为合成型平滑肌细胞更容易参与炎症、基质降解和血管壁变弱。敲低B细胞中的LUM后,这种促转换作用明显减弱;相反,过表达LUM则进一步增强平滑肌细胞向合成表型转变。
通路分析也支持这一机制。CD79A主要富集于TNF信号、B细胞受体信号、C型凝集素受体信号和炎症反应;LUM则与NF-κB信号、补体、炎症反应、凝血、血管生成、上皮—间质转化以及细胞外基质重塑相关。LUM还与COL3A1呈显著正相关,进一步提示其可能参与胶原和血管壁结构重塑。研究还进行了分子对接和分子动力学模拟,显示LUM与白藜芦醇、CD79A与苯并芘可形成相对稳定复合物,为未来小分子干预提供了初步结构线索,但作者也强调这仍属于计算预测,需要实验验证。
这项研究提出了一个新的AAA进展模型:在腹主动脉瘤组织中,B细胞异常富集并被激活,其中LUM阳性B细胞通过炎症通讯和旁分泌作用影响血管平滑肌细胞,使其失去正常收缩功能、转向促重塑的合成状态,最终加速血管壁结构破坏和动脉瘤扩大。该研究不仅加深了人们对AAA免疫微环境的认识,也把LUM阳性B细胞推向了潜在诊断标志物和治疗靶点的位置。未来,如果能在更大临床队列和动物模型中验证这一机制,针对LUM或特定致病B细胞亚群的精准干预,或许将为腹主动脉瘤患者带来非手术治疗的新希望。

Nature | 单细胞揭示TROP2高表达驱动结直肠癌转移与化疗耐药,化疗联合 ADC 实现协同增效
如果你对单细胞转录组研究感兴趣,但又不知道如何入门,也许你可以关注一下下面的课程


看完记得顺手点个“在看”哦!
 生物 | 单细胞 | 转录组丨资料
生物 | 单细胞 | 转录组丨资料长按扫码可关注
