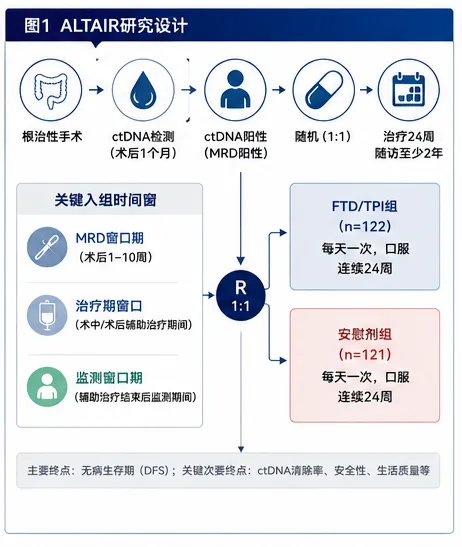
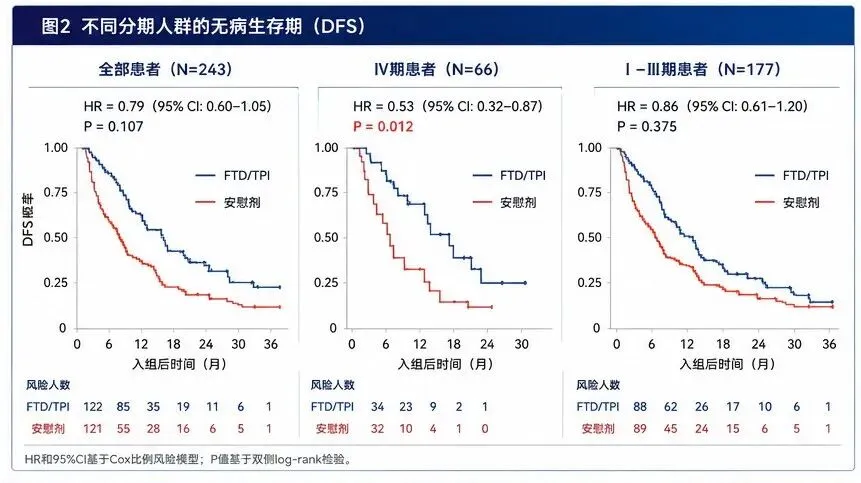
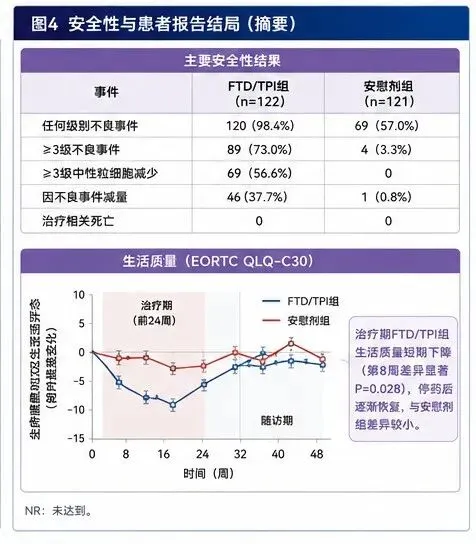
耿同学讲的那些故事,很多人应该都看到了。他在生物医学圈里掀起来的那场打假风暴,P图、Western blot裁切、数据矛盾、图片复用,一篇文章一篇文章地撕,真的很猛。但是在真正的临床一线,最害人的东西还不是P图的基础研究。真正难以分辨的,是那些数据全是真的、分析也是规范做出来的,但结论的"气氛"被推到了它本不该到的位置的临床论文。一堆看起来完美无瑕的III期临床试验,数据真实、图表漂亮、经过了peer review,发出来的时候金光闪闪,临床医生看了以后也跃跃欲试。而这每一个决策背后,都是一个活生生的人。所以我搭了一套专门用来审临床试验论文的系统,并且给它起了个名字,叫"临床研究照妖镜"。今天拿一篇论文,现场演示一下这套东西是怎么工作的。被拆解的论文前几天,Nature Medicine发了一篇III期临床试验的论文,叫《Post-adjuvant chemotherapy in ctDNA-positive patients with resected colorectal cancer》。简单说,就是在大肠癌患者做完手术和标准化疗之后,用血液中的循环肿瘤DNA来监测有没有残留的癌细胞。如果检测出来ctDNA是阳性的,说明体内可能还有微小的残余病灶,但CT上还看不见。这时候,一个很直接的想法就冒出来了,我能不能提前用药干预?在CT还看不到病灶的时候就下手,会不会比等影像学确认复发再进行治疗效果更好?这篇论文就是为了回答这个问题,试验代号叫ALTAIR。研究者找了一种药,叫朗斯弗,学名是FTD/TPI。这种药的有效性在晚期肠癌里是被验证过的,所以他们想试试看,把它提前用在ctDNA阳性但影像学还没复发的患者身上,能不能改善结果。试验设计很标准,随机双盲、安慰剂对照。243个术后I~IV期的肠癌患者,一半吃药一半吃安慰剂,随访了差不多两年。这里要提前说明一句:入组的门槛其实不是"分期",而是"根治性切除 + 影像学无病灶 + ctDNA阳性"这三条。所以里面确实有66个IV期患者(占24.2%)——但都是原发灶和转移灶都做了根治性切除、CT上已经看不到任何病灶的寡转移患者。研究的主要终点DFS,也就是无病生存期,中间值是9.30个月对5.55个月,HR=0.79,P=0.107。在临床试验的统计学标准里,0.05是门槛。P值大于0.05,就等于统计学上没有显著差异,而0.107比0.05大了一倍多,也就是主要终点,没达到。但是,论文的作者在讨论部分开始了一段非常值得玩味的叙事编排。他们开始大量强调几个所谓的探索性分析,比如说,在IV期患者这个亚组里,HR达到了0.53,P=0.012。再比如说,他们做了一次事后的中心影像审查,把几个病例从“复发”改判成了“没复发”之后,重新算了一遍,P值变成了0.0406。好了,我知道你可能在想,这个结论到底能不能信?这就要说到“临床研究照妖镜”了。什么是“临床研究照妖镜”它不是一个普通的审稿工具,更不是一个单纯的摘要生成器。它是带着预设怀疑进来的,这可能是它跟所有其他审稿系统最本质的区别。标准的学术审稿流程,是拿到一篇论文,先读背景,再读方法,然后看结果,最后对照讨论和结论,判断它是不是合理。这里面有个问题,你从一开始就是用“作者视角”在读,你在跟着他的思路走。而照妖镜的做法刚好相反。它有一个叫“反直觉审查协议”的东西,拿到论文的第一件事,不是去思考这篇文章的结论有多靠谱,而是让你先写一段,为什么这篇文章可能完全不可信。你得先把所有怀疑列出来,然后再去找证据证伪它。它有一个完整的六步流程。第一步:出身核查先别管数据好不好看,先看看这篇论文是谁出的钱,通讯作者跟药企有什么关系,注册信息有没有被改过。这一步大概花五分钟,很多时候五分钟就够判断一篇论文的底色了。第二步:设计审查用的是RoB 2.0这个工具,六个维度去评估偏倚风险。随机化是怎么做的,分配隐藏了没有,盲法执行了没有,脱落率怎么样,有没有选择性报告。还有一条很多人会漏掉的——纳入人群到底是不是同质的,试验里的干预有没有偏离指南和临床原则。第三步:统计硬核审查这是照妖镜的心脏。它有一个统计学十问,不是只看P值有没有小于0.05。它要看你用的是什么统计模型,你的比例风险假设成不成立,ITT和PP分析的结论是不是一致的,ARR换算成NNT是多少,多重比较有没有校正,亚组分析做了交互检验没有。第四步:反药企陷阱检测这可能是整个系统里最独特的一个模块。它预设了四种药企最常用的操纵模式,一种一种地去检。非劣效性的边界有没有放水,复合终点是不是由不重要的组分驱动的,亚组是不是在P-hacking,中期分析有没有过早叫停。它还有一个叫「魔鬼辩护人」的角色,专门站在药企律师的视角去写最强辩护词,然后我再一条一条驳回去。第五步:药物安全性交叉验证毒副反应数据是不是完整的,有没有把最严重的副作用藏到补充材料里了,生活质量数据有没有选择性报告。第六步:文献定位这篇文章在整个治疗格局里到底处在一个什么位置,跟同类的III期研究对比,真有增量价值吗。这六个步骤,光看清单的话好像挺工整的,但实际跑起来的时候,它内部是模拟了四个审查官在同时工作的。一个是临床主编,看整体结构。一个是统计审查员,抓数据真实性。一个是安全监察员,专门盯毒性。还有一个,就是刚才说的魔鬼辩护人,这个角色是最好玩的,也最让人头疼,因为他发现的问题,按系统的铁律,必须进最终报告,而且如果他认定核心结论不可信,这篇论文的评级最高只能到“存疑”。然后系统会给论文打一个可信度评级,从 ⭐⭐⭐⭐⭐ 到 ⭐。这套东西的核心原则就是一句话,有预设怀疑倾向的建设性批评。我不是中立观察者,我是代表患者和临床实践利益的独立审判者。对ALTAIR论文的拆解过程回到ALTAIR这篇论文,照妖镜的出身核查一跑,第一张红灯就亮了。这篇文章里,Taiho出钱出药,然而离谱的是,提供ctDNA检测的Natera公司,有六个员工挂在作者列表里,通讯作者的COI声明打出来有一页半。有人说,这很正常啊,药企资助的III期试验都是这样,而且论文白纸黑字写了这是研究者发起(investigator-initiated)、资助方"没有参与研究设计和分析"。对,这确实很常见,但常见不等于没问题,声明写了不等于就能免疫。一页半的COI声明,翻译过来就是,几乎每一个参与这个试验的关键决策者,都同时在拿着另一只手从药企领钱。你可以说它形式上独立,但在利益结构上,它更像一次利益共生的联合路演。这一条我不把它当成铁证,只当成一盏提醒你后面每个数字都要多看两眼的黄灯。出身核查之后,照妖镜进入了红旗信号快速扫描。系统内置了10个红旗,包括赞助方深度介入、主要终点阴性、讨论中大量强调事后分析、分层因素事后修改、PH假设违反、极端毒性差异等等。每命中一个亮一个灯,ALTAIR这篇论文,命中了8个。任何一个在严谨的审稿里,单独拎出来都够审稿人发一条critical comment的。但它就这样,完好无损地过了Nature Medicine的peer review。在进入统计之前,设计审查那一步,照妖镜其实先亮了一盏更根本的红灯——关于"这群病人到底是谁"。这243个人,是从三个完全不同的窗口进来的,而这三类人跟"标准辅助化疗"的关系,天差地别。第一类,监测窗口,152人,占62.6%,是最大的一块。这些人做完了全部该做的标准治疗,该上奥沙利铂双药的都上完了,影像学阴性,之后在随访里ctDNA转阳才入组。对这类人,现行指南根本没有推荐任何治疗——ctDNA阳性但CT阴性,标准做法就是观察。所以对这63%的人,安慰剂对照恰恰就是当前标准,谈不上违反指南。真正的问题在另外两类。MRD窗口,58人,占23.9%,是术后2到10周、辅助化疗还没开始就ctDNA转阳入组的。对一个III期病人,标准是奥沙利铂双药辅助,这时候把他放进一个可能吃FTD单药或安慰剂的试验,本身就存在用低强度方案顶替标准双药的风险。on-treatment窗口,33人,占13.6%。这些人是正在做辅助化疗期间ctDNA转阳的,而论文明确要求——"ACT was required to be discontinued prior to initiation of study treatment"——也就是主动停掉正在进行的FOLFOX,换成FTD单药。对一个III期病人来说,这是最直接的治疗降级。IIB到III期术后,标准就是奥沙利铂加氟尿嘧啶双药,把病人从双药上撤下来,换成一个从未被证明有辅助获益的低强度单药,这在临床原则上是说不过去的。而最漂亮的一击,是论文自己的数据把这个问题坐实了。on-treatment窗口的病人,FTD/TPI组反而更差,HR=1.15(95% CI 0.54–2.44,Extended Data Fig 3B)。论文讨论里也白纸黑字承认:"no benefit—and a numerically worse DFS—was observed among patients enrolled after ctDNA positivity during ACT。"换句话说,被从奥沙利铂双药上撤下来、改用FTD单药的那一组,结局是往坏里走的。所以这里真正的问题,不是"整个试验用低强度单药违反指南"这么一刀切——对63%的监测期病人这话不成立——而是:这个试验把三类临床上根本不该混为一谈的人群,尚未辅助的、辅助中途的、辅助完成后监测的,合并进了同一个主要终点。人群异质性叠加治疗降级,主要终点阴性,一点都不意外。到了统计硬核审查这一步,才是真正让我震惊的部分。统计学十问的第三问,PH假设检验。简单解释一下,当你用HR来衡量两组之间的生存差异时,你背后有一个前提,就是药物效应在整个随访期内是恒定的。如果这个前提被推翻,如果药效在前三个月很强但后面就没了,那HR作为一个汇总统计量就失效了。ALTAIR的Schoenfeld残差检验结果是P=0.0027,PH假设被明确推翻。翻论文里的KM曲线,曲线自己就已经讲完了整个故事。6个月的时候,FTD/TPI组的无病生存率是70.5%,安慰剂组是45.5%,差了25个百分点。但到12个月,FTD/TPI组掉到了31.8%,安慰剂组26.8%,差距缩到了5个百分点。到18个月,两条线基本重合(0.21对0.22,FTD/TPI组甚至略低)。到24个月,FTD/TPI组16.9%,安慰剂组14.5%,几乎一样。这不是治愈。这更像是把做CT检查、确认复发的时间往后推了两个多月而已。然后是那个最引人注目的操作,事后中心影像审查。论文的主要终点评定是由研究者自己做的。每个患者有没有复发,由研究站点自己判断,这个判断本来就有主观成分。有些病灶到底是不是复发,不同人看可能会有不同结论。于是论文在数据库锁定之后,额外委托了一个四人放射专家小组,对所有的影像进行盲态复阅。复阅的结果是,243个患者里,有10个的判断跟研究者不一致。其中6个是研究者判为复发但专家判为没复发,4个反过来。然后他们用专家改完的结果重新算了一遍DFS,P值从0.107变成了0.0406。在243个人的试验里,是4.1%。4.1%的改判,就让整篇论文从“不显著”变成了“显著”。虽然作者本人并没有拿0.0406去宣称翻盘,但是挡不住这个数字的下游命运。一旦这篇论文进入流通,"P=0.0406、有显著获益"这句话就会被从上下文里抠出来,被拿去跟医生讲,被快速阅读的人当成结论记住。然后是安全性审查,照妖镜的安全监察员开始干活了。FTD/TPI组,发生了至少1次3级或以上不良事件的患者比例是73%,而安慰剂组是3.3%。73%对3.3%,这是质的不同。而且95%的患者在治疗期间至少跳过一次剂量,几乎每个患者都是拖着残血在扛这个方案。而他们的QoL,生活质量评分,在治疗期间是显著恶化的。week 8的时候,Global Health Status的P值是0.028。然后讨论里写什么,说这种恶化是暂时的、停药后就恢复了,某些功能量表还显示了一定程度的改善。一个主要终点阴性的试验,用73%的3级毒性、95%的剂量跳过、治疗期显著恶化的生活质量,去换一个统计学上不成立的获益——这笔账,无论怎么算都是亏的。然后到了反药企陷阱检测模块,这篇论文命中了四大操纵模式中的两个。第一个,亚组P-hacking。主分析没显著,就开始挖亚组。IV期患者这个亚组HR=0.53,P=0.012,在结果和讨论里被反复强调,每次都用"探索性"这个词打掩护。问题是,多重比较没做校正,论文自己也承认了,而且分期的交互作用P值根本没报。这里补一刀,让这个质疑更致命:这66个IV期病人,恰恰是全队列里基线ctDNA负荷最高的,中位MTM/ml是0.68,而I、II、III期只有0.19、0.29、0.38。论文自己也承认"基线分子负荷越高、表观获益越大"。同时它又把"高解剖负荷、影像学已经复发"的病人排除在设计之外。所以IV期那个HR=0.53的信号,是被基线ctDNA负荷严重混杂的——它确实是真的IV期病人,但"IV期特别获益"这个因果解释根本立不住。既没做分期交互检验,又有明显混杂,凭什么把这个亚组当亮点?第二个,安全性信号弱化。刚才说的QoL数据,选择性报告不好的部分,强调非核心的改善点,这就是操纵。最后是魔鬼辩护人环节。我让它站在药企律师的角度,写这篇论文的最强辩护词,它是这么写的:ALTAIR是第一个完成入组的TOMR策略III期试验,这个历史意义本身就值得发表。主要终点虽然没达到,但趋势是正向的,HR=0.79的方向是获益的。亚组分析虽然是探索性的,但提供了一个重要的假设生成信号,可以为后续研究提供方向。事后中心审查虽然是锁库后追加的,但盲态执行规范,kappa值高达0.866,说明判读质量是稳定的。安全性方面,毒性谱是已知的,跟晚期适应症一致,没有新的安全性信号,临床上是可以管理的。听起来好像有点道理?然后照妖镜一条一条驳回去。第一,PH假设P=0.0027,也就是说HR=0.79这个"趋势"本身就是虚假的,前几个月有效不代表整个随访期有效,24个月的时候作者自己算的HR都到0.921了。用一个已经失效的汇总统计量去论证"方向是正向的",是统计学上的严重错误。第二,事后中心审查改变了P值,但这本身就不是预设分析。你没法用一个非预设的结果来修复一个预设分析的失败。这就像考试不及格之后,去找老师重新判卷,只改了10道题就及格了,你不能说这10道题本来就应该这么判。作者自己也承认它不改变主结论,那它就更不该成为任何人记住的那句话。第三,73%的3级及以上毒副反应率,对于主要终点阴性的试验来说是不可接受的代价。如果主要终点都证明不了获益,那任何3级及以上的毒性都是多余的伤害。更何况获益风险比根本不成立,RMST只多出1.93个月,95% CI是 −0.95到4.82,P=0.189。在统计学上,这甚至不是一个显著的数字。第四,人群本身就不该被这样合并。on-treatment那一组被从标准双药上撤下来换单药,结果HR=1.15,往坏里走。一个把III期病人从指南方案上撤下来、还让他们更差的设计,不该被"uniform intervention"这四个字洗白。最终判定,可信度评级给了 ⭐⭐。高风险,不建议采信核心结论。拆这篇论文的意义回到更大的问题,Nature Medicine的peer review为什么没拦住?这不是个别的审稿人失误。这是一个系统性的激励结构问题。药企花了几千万甚至上亿美金做完一个III期试验。如果是阴性结果,能不能发Nature Medicine?大概率不行。说句公道话,这篇论文的讨论和局限性写得其实相当坦白,PH违反、疗效衰减、RMST不显著、中心审查是探索性的、毒性沉重、OS数据不成熟——这些作者基本都自己招了。所以它不是那种赤裸裸造假、或者把数据藏起来的论文。但也正因为如此,它才更值得拆。因为最难防的,不是把坏数据藏起来,而是把所有坏数据都诚实地写进讨论和补充材料的深处,同时把最乐观的那个版本——0.79、0.53、0.0406——放进摘要、正文和图表标题这些最显眼的位置。一篇论文可以在字面上句句属实,同时在气氛上系统性地乐观。临床医生记住的,是那几个被放大的数字,不是补充材料里的P=0.189。耿同学打假,撕的是数据造假。而临床研究照妖镜的工具是统计学和逻辑推理,它撕的不是造假,是合法的、诚实的、却把结论气氛推过了头的包装。这两件事,殊途同归。在过去的几年里,我看到很多类似的临床试验文章,数据是真实的,图表是漂亮的,方法也是标准的,局限性甚至都写得很诚实,但结论的重心被悄悄挪到了一个它本来不该到的位置。所以我才搭了这个体系,不是为了表演什么。是因为我需要一个不被药企叙事、也不被"讨论写得很诚实"这层表象带走的判断工具。后续我打算把它做成一个可复用的AI工作流,让更多的临床医生能拿它来审看到的论文,而不是只有我自己在用。耿同学的事情让我意识到,可能有更多的医生需要这个东西。后续如果对这套“临床研究照妖镜”的工作流程感兴趣的话,可以后台给我留言,我可以把这个工具分享给你。
基本文件流程错误SQL调试
请求信息 : 2026-07-09 13:45:27 HTTP/1.1 GET : https://www.yeyulingfeng.com/a/843836.html
 夜雨聆风
夜雨聆风