夜雨聆风
夜雨聆风
在医疗器械质量管理体系中,DMR(Device Master Record,器械主记录)是一份至关重要的文件。它明确规定了产品的生产、测试、包装、标签和放行流程,相当于医疗器械的"生产配方"或"制造蓝图"。DMR的完整性和准确性直接关系到产品能否合规生产、顺利通过审核。
医械加油站结合FDA QSR 820、ISO 13485:2016及欧盟MDR的法规要求,系统拆解DMR的核心内容、与DHF/DHR的区别、与风险管理的关联、变更控制及发放管理,帮助企业建立合规、可操作的DMR体系。
法规定位:FDA QSR 820.181明确要求每类器械必须建立DMR;ISO 13485:2016中虽使用"医疗器械文档(MDF)"概念,但实质内容与DMR基本一致。在FDA QMSR新规(与ISO 13485对齐)背景下,DMR的实质要求并未降低。
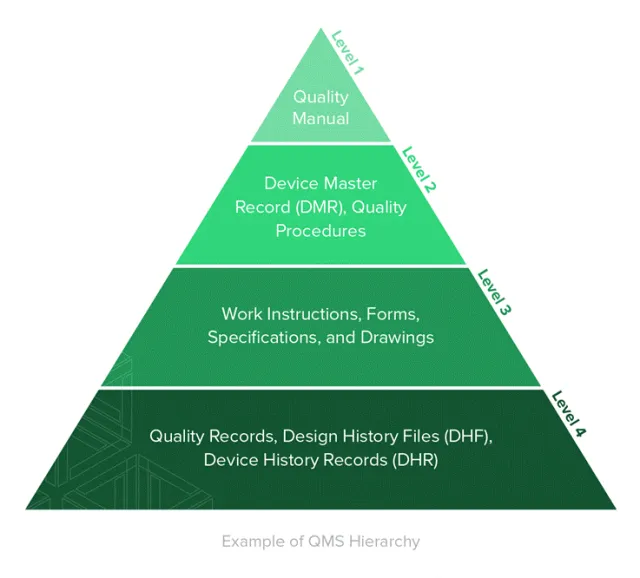
质量管理体系层级——DMR位于Level 2核心层
一、DMR与DHF、DHR的区别
DMR、DHF、DHR是医疗器械质量管理体系中三个最容易混淆的概念,也是审核过程中最常出现问题的环节。
三者的核心区别在于各自回答的问题不同、所处的生命周期阶段不同。
文件 | 全称 | 核心作用 | 生命周期 |
DHF | Design History File 设计历史文件 | 记录设计过程 | 研发阶段 |
DMR | Device Master Record 器械主记录 | 定义制造规范 | 设计转换→量产 |
DHR | Device History Record 器械历史记录 | 记录实际生产 | 生产阶段 |
三者的关系链条清晰明确:DHF输出设计结果,DMR吸收最终设计和转产要求形成制造蓝本,DHR逐批记录实际生产是否按DMR执行。
审核员通常会选择一款已上市设备,按照"DHF → DMR → DHR"的路径逐一检查。如果DMR与当前设计不符,或DHR中显示的装配顺序、测试方法与DMR不一致,则表明设计控制、文档控制和生产控制存在脱节。
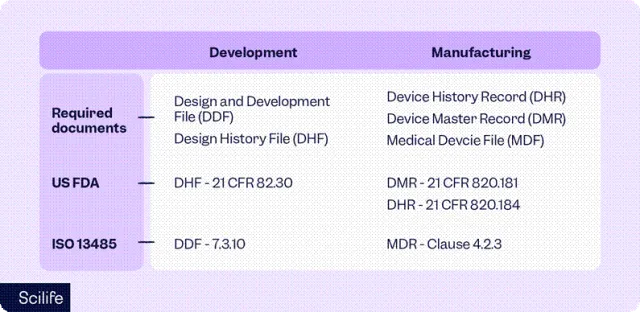
图DHF、DMR、DHR在法规体系中的对应关系
二、DMR的核心内容构成
根据FDA 21 CFR 820.181和行业实践,DMR应包含或引用以下五大类信息。企业可按此框架逐项核对,确保DMR内容完整、无遗漏。
类别 | 包含内容 | 回答的问题 |
产品规范 | 图纸、BOM、组件规范、软件规范 | 产品是什么 |
生产过程规范 | 设备规范、生产方法、工艺流程、环境要求 | 如何制造 |
质量保证规范 | 检验规程、接收准则、检测设备 | 如何检测 |
包装和标签规范 | 包装方法、标签内容、UDI载体 | 如何包装标识 |
安装维护服务规范 | 安装方法、维护程序、维修指南 | 如何使用维护 |
产品规范
产品规范是DMR的基础层,回答"产品是什么"的核心问题。
应包含:产品图纸(原理图、零件图、部件图、总装图,含尺寸公差和技术要求)、物料清单BOM(物料编码、规格型号、材质牌号、供应商信息)、组件规范(外协件/标准件/自制件的验收标准)、软件规范(软件版本、配置要求)、产品技术要求(型号规格、性能指标、检验方法)。
生产过程规范
生产过程规范回答"如何制造"的问题,是DMR中内容最丰富的部分。
应覆盖:工艺流程图(含关键工序和特殊过程标识)、作业指导书/工艺卡(操作步骤、温度/压力/时间等工艺参数)、设备规范(设备清单、操作规程、校准状态)、工装夹具规范、生产环境规范(洁净度、温湿度)、清洁规范(清洗参数、清洁剂、验证记录)。
质量保证规范和程序
质量保证规范回答"性能指标是什么、如何检测"的问题。
应包含:进货检验规程、过程检验规程、成品检验规程、检验记录表格、检测设备清单、过程控制图、返工/返修规程。各项检验规程必须覆盖产品技术要求的全部性能指标,确保数据可追溯。
包装和标签规范
包装和标签规范直接关联法规合规性。
应包含:包装图纸(初包装和中包装的尺寸规格)、标签规范(内容、尺寸、位置、材质,符合《医疗器械说明书和标签管理规定》)、UDI数据(DI/PI编码规则,符合GS1等发码机构标准)、UDI载体规格、贴标签程序(含防差错措施)、灭菌/有效期要求(包装完整性验证、货架有效期)。
安装、维护及服务程序
对于需要安装、维护或售后维修的产品,
DMR还应包含:安装程序(安装方法、调试步骤)、维护程序(校准方法、消毒再处理流程、维护周期)、服务程序(维修方法、故障排查指南)、储存和运输规范。
组织形式建议:DMR不需要收集所有资料进行复印汇总,可以通过文件清单索引模式(DMR List/Index)指明相关文件的位置和版本,便于查找和维护。
三、DMR与风险管理的关联
DMR不仅是制造规范,更是风险管理落地的关键载体。ISO 14971中确定的风险控制措施,必须通过DMR逐项落实到具体的规格、工艺或程序中。
风险控制类别 | DMR中的体现方式 |
设计风险控制 | 转化为设备规格、产品图纸、材料要求 |
制造风险控制 | 转化为工艺参数、检验计划、过程控制 |
使用信息控制 | 转化为标签、使用说明书(IFU) |
当DMR与风险文件一致时,审核员可以完整追溯每个风险控制措施:从危害识别 → 设计决策 → 具体要求或流程步骤 → DHR中的客观证据。成熟的企业会定期将DMR内容与风险管理文档进行核对,确保两者在变更累积过程中保持同步。
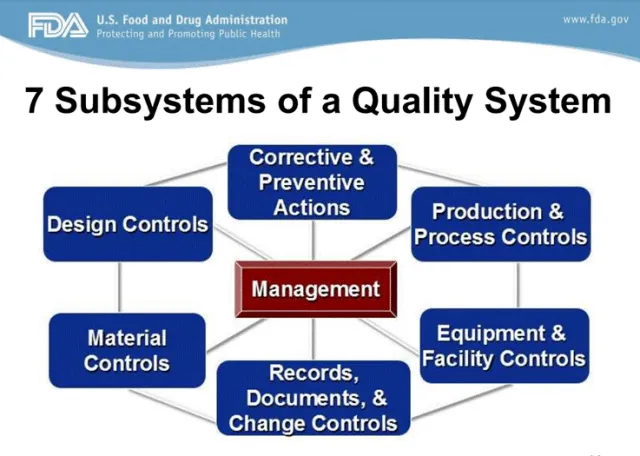
FDA质量体系的7大子系统——记录、文件与变更控制是核心之一
四、DMR的变更控制
DMR界定了产品可以合法上市的依据,对其进行的任何更改都必须经过正式审批流程。变更管理是DMR维护中最容易被忽视的环节,也是审核中的高频不符合项。
触发DMR变更的常见情形
设计变更:图纸修订、物料变更、软件版本升级
工艺优化:工序调整、参数修改、设备更换
测试方法变更:检验方法修订、标准更新
标签变更:标签内容调整、UDI信息更新
变更审批路径
因设计变更(图纸、物料等):开发设计部更新DMR文件 → 开发设计部负责人审核 → 管理者代表批准。
因工艺优化(工序、作业方法等):生产部提出变更需求 → 开发设计部与生产部共同审核 → 管理者代表批准。
在质量体系中,通常通过设计变更控制程序来管理DMR的更新。变更后需同步评估对DHF、DHR的影响,确保三类文件的一致性。
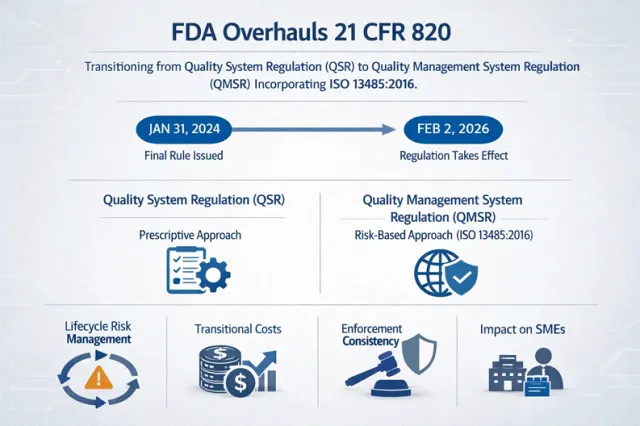
FDA从QSR向QMSR过渡——DMR管理要求与ISO 13485进一步对齐
五、DMR的发放与管理
发放类型
内部发放:经授权发放给各职能部门用于作业、管理、核查
外部发放:因工作需要提供给供方、监管机构、认证机构等,外发应单独留档以便核对
管理要点
索引而非复印:通过文件清单索引模式指明相关文件的位置,便于查找和维护
版本管理:发生设计变更或工艺变更时,同步维护和更新DMR中的相关内容
外包场景:法定制造商仍是DMR的最终所有者,必须在质量体系内定义、审批和控制,根据质量协议向合同制造商提供受控副本
六、各阶段文件清单速查
为便于企业快速定位各阶段的核心文件,医械加油站整理以下速查表:
阶段 | 文件类型 | 示例内容 |
研发 | DHF | 设计输入、输出、验证、确认、评审、变更记录 |
设计转换 | DMR | 图纸、BOM、工艺规程、检验规程、标签规范 |
生产 | DHR | 生产记录、检验记录、放行记录、UDI赋码记录 |
DMR是医疗器械从设计到量产的关键桥梁,其完整性直接影响产品的合规生产和审核通过率。建立一套清晰、完整、可追溯的DMR体系,不是应付监管的形式工作,而是保障产品质量、降低合规风险的核心能力。
医械加油站将持续输出医疗器械质量管理体系、设计开发、合规注册等实操干货。如有DMR搭建或审核方面的疑问,欢迎评论区留言交流。