 夜雨聆风
夜雨聆风





【干货分享】风险管理文档
风险管理文档
产 品 名 称 :
编 制: 日 期:
批 准: 日 期:
风险管理文档目录
1 、风险管理计划
1.1目的
1.2范围
1.3产品基本信息
1.4职责和权限的分配
1.5风险可接受性准则
1.6风险控制
1.7产品各生命周期中的风险管理活动
1.8风险管理活动评审的要求
1.9风险管理活动验证的要求
1.10生产和生产后信息的收集
2、风险分析
2.1概述
2.2风险分析人员
2.3医疗器械预期用途和与安全有关特征的识别
2.4初始危险的判断及初始风险控制措施表
3、风险评价
3.1概述
3.2风险评价记录
3.3风险评价结果
4、风险控制措施的实行和验证记录
4.1概述
4.2风险控制方案分析
4.3风险控制表
4.4风险控制的验证评审记录
5 、综合剩余风险
5.1 概述
5.2 综合剩余风险的可接受评定
6、 风险管理报告
6.1 概述
6.2 风险管理评审输入
6.3 风险管理评审
6.4 风险管理评审结论
1.风险管理计划
1.1目的
本风险管理计划为干式荧光免疫分析仪(型号FIC-H1)的风险管理活动提供系统的方法,为风险管理提供指南,明确管理的目标,防止遗忘重要的内容。
1.2范围
本风险管理计划主要是对上述产品在其整个生命周期内(包括可行性分析阶段、设计开发阶段、生产阶段、销售安装阶段、使用和上市后阶段等)进行风险管理活动的策划
1.3产品基本信息
1.3.1产品描述
A概述:
干式荧光免疫分析仪属于临床检验仪器类医疗器械,与适配的基于免疫层析法的特定干式试剂配套,供人体样本的免疫检测用。属于6840-3免疫分析系统仪器。
B产品组成:产品由仪器和电源适配器组成.
C 预期用途:
本仪器与适配的基于免疫层析法的特定干式试剂配套,供人体样本的免疫检测用。
D 预期目的:
通过使用仪器配套试剂对人体样品的临床指标进行,提供相应的临床检验报告。
E 适用环境:
干式荧光免疫分析仪可用于医疗机构的中心实验室、门/急诊化验室、临床科室和其他医疗服务点(如社区医疗点)、体检中心等,也适用于科研实验室。
F产品预期使用寿命 :5年
1.3.2参考标准
1、GB 4793.1-2007《测量、控制和实验室用电气设备的安全要求 第1部分:通用要求》
2、GB 4793.9-2013 《测量、控制和实验室用电气设备的安全要求 第9部分实验室用分析和其他目的自动和半自动设备的特殊要求》
3、YY 0648-2008 《测量、控制和实验室用电气设备的安全要求 第2-1014、部分:体外诊断(IVD)医用设备的专用要求》
4、GB/T 14710-2009 《医用电器环境要求及试验方法》
5、GB/T 18268.1-2010 《测量、控制和实验室用的电设备 电磁兼容性要求 第1部分:通用要求》
6、GB/T 18268.26 《测量、控制和实验室用的电设备电磁兼容性要求 第26部分特殊要求 体外诊断(IVD)医疗设备》
7、YY/T 0316-2008 《医疗器械风险管理对医疗器械的应用》
8、YY/T 0466.1-2016《医疗器械用于医疗器械标签、标记和提供信息的符号 第1部分:通用要求》
1.4职责和权限的分配
总经理为风险管理提供适当的资源,对风险管理工作负领导责任。保证给风险管理、实施和评定工作分配的人员是经过培训合格的,保证风险管理工作执行者具有相适应的知识和经验。
研发部负责产品设计和开发过程中的风险管理活动,形成风险分析、风险评价、风险控制、综合剩余风险分析评价的有关记录,并编制风险管理报告。
质量部、销售部、生产部等相关部门负责从产品实现的角度分析所有已知的和可预见的危险以及生产和生产后信息的收集并及时反馈给研发部进行风险评价,必要时进行新一轮风险管理活动。
研发部和评审组成员定期对风险管理活动的结果进行评审,并对其正确性和有效性负责。
办公室负责对所有风险管理文档的整理工作。
1.5风险可接受性准则
生产部、质量部、销售部负责配合研发部对经风险分析判断出的危险进行发生概率与损害严重度的分析,最后根据本计划确定的风险可接受准则判断风险的可接受性,保存好评价记录。以下是为本次风险管理确定的风险可接受准则,其中损害的严重度采用定性分析,损害发生的概率采用半定量分析,风险可接受性准则以5Ⅹ5三分区矩阵图表示。
1.5.1严重度准则

1.5.2发生度准则

1.5.3风险可接受性准则

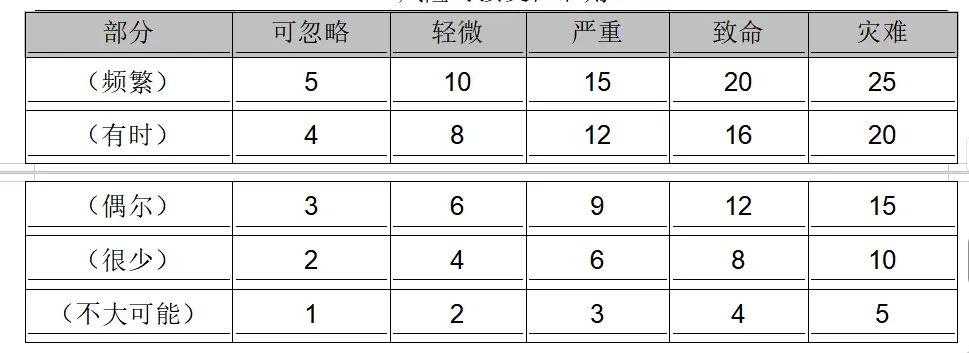
风险分数的种类
风险可接受区(ACC) 风险等级: 1~4 该区域内风险可接受
风险合理降低区(ALARP) 风险等级: 5~12 该区域内形成的风险应采取适当的措施移除风险或降低风险,然后评估总体剩余风险。采取适当的措施后,剩余风险若不可授受的,应将医疗受益与剩余风险作比较。若预期使用的医疗受益超过剩余风险,则可以接受。否则应进行设计和开发变更或停止研发。
风险不可接受区(NACC) 风险等级: 15~25 采取措施移除风险或降低风险后,若剩余风险仍处于“不可接受区”,则进行风险/受益评价。
在经过风险分析和风险评价过程判断出的产品所有的风险均应采取合理可行的措施降至可接受区,当风险被判断为不可接受时,应应收集相关资料和文献对风险进行风险/受益分析,如果受益大于风险,则该风险还是可接受的,如果风险大于受益则设计应放弃。
对损害概率不能加以估计的危险处境,应编写一个危险的可能后果清单以用于风险评价和风险控制,各部门应配合技术部采取合理可行降低法将风险降低到合理可行的最低水平,对于无法降低的风险进行风险/受益分析,如果受益大于风险,则该危险可接受,如果风险大于受益,则风险不可接受。
在可接受区,风险是很低的,但是还应主动采取降低风险的控制措施。
受益必须大于风险才能判断为可接受。
1.6风险控制
对于经判断为可接受的风险还应当采取可行的措施将风险降到最低。
对于经判断为不可接受的风险,各部门应配合技术部在设计开发阶段从以下几个方面进行风险控制方案分析,识别一个或多个风险控制措施,以把风险降低到可接受水平。
1)用设计方法取得固有安全性
—消除特定的危险;
—降低损害的发生概率;
—降低损害的严重度。
2)在产品本身或在制造过程中的防护措施。
3)安全信息
—在产品随附文件中给出警告、使用说明;
—限制医疗器械的使用或限制使用环境;
—对操作者进行培训。
在产品试生产或生产阶段,对产品制造过程进行控制。
如果经方案分析确定所需的风险降低是不可行的,则各部门应收集相关资料对剩余风险进行风险/受益分析,若经评审所收集的资料和文献不支持受益大于风险,则设计应放弃。
各部门应确保经判定的危险处境产生的一个或多个风险得到了考虑,保证风险控制的完整性。
在风险控制方案实施中或实施后,应对实施效果进行验证,以确定控制措施的适应性和有效性,对任何剩余风险都应采取本计划中第4条的风险可接受准则进行评价,对判断为不可接受的应采取进一步的风险控制措施,如果控制措施不可行,则应收集和评审相关的资料和文献对剩余风险进行风险/受益分析,若受益大于风险,则剩余风险依然是可接收到,如果风险大于受益,则为不可接受。对于判断为可接受的剩余风险,外贸部、销售部应配合技术部决定那些剩余风险应予以公开,依据YY/T0316:2008附件J的指南公开哪些剩余风险。同时对控制措施的实施是否会引起的一个或多个新的风险或对采取措施之前评价的风险是否有影响进行分析,必要时进行再次风险分析、风险评价和风险控制,所采取活动的结果应进行记录并保持,此过程预期30个月。
1.7产品各生命周期中的风险管理活动
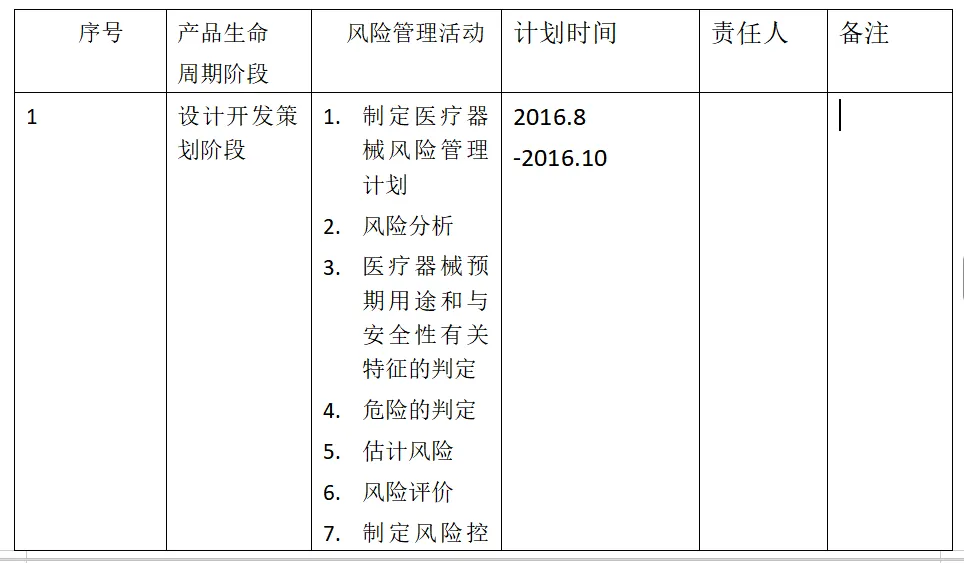
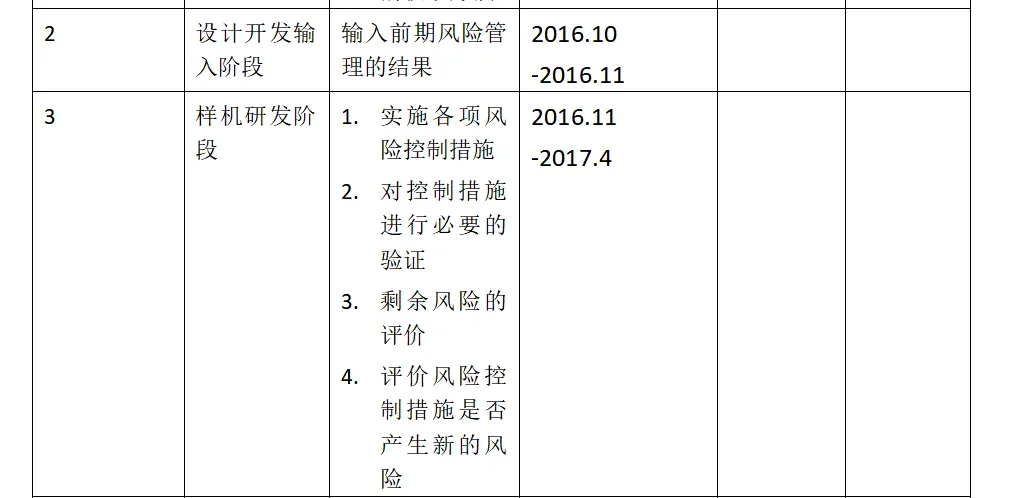
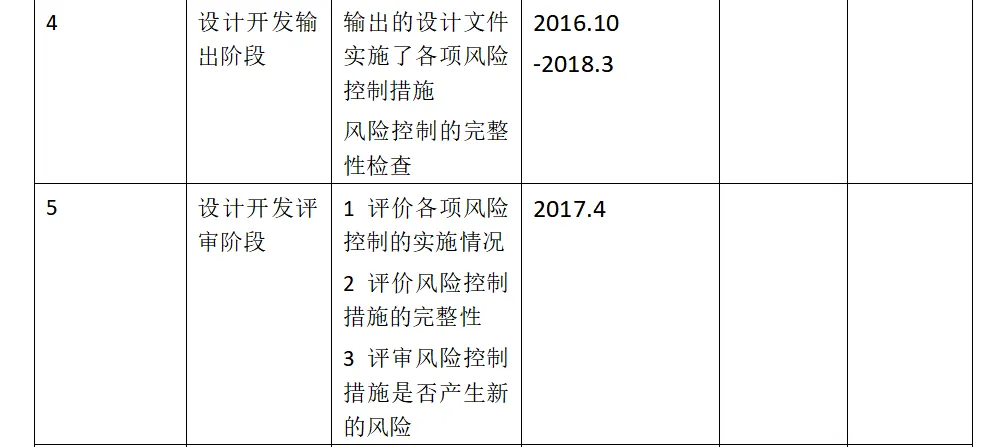
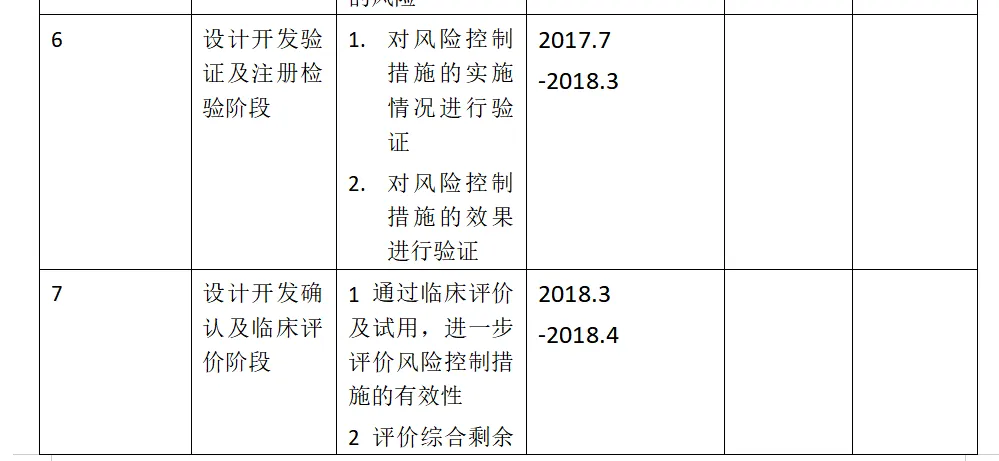
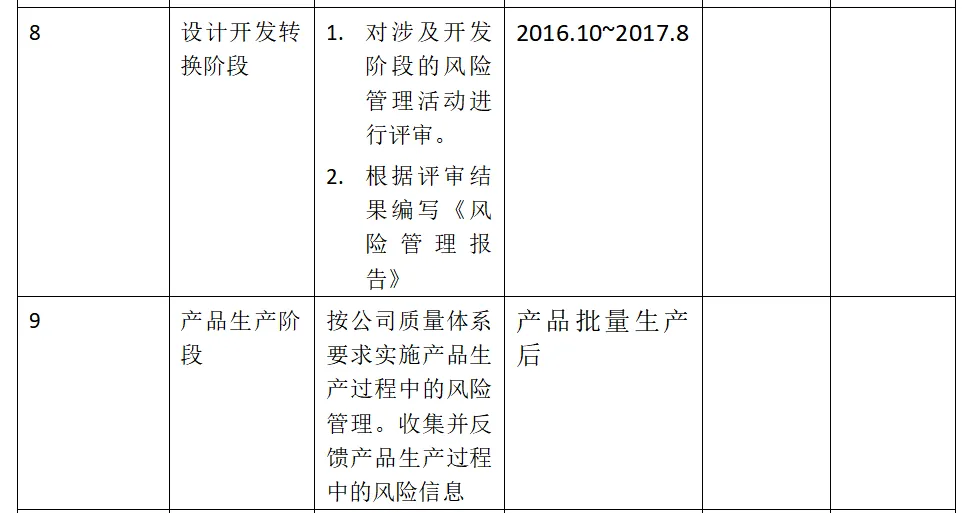
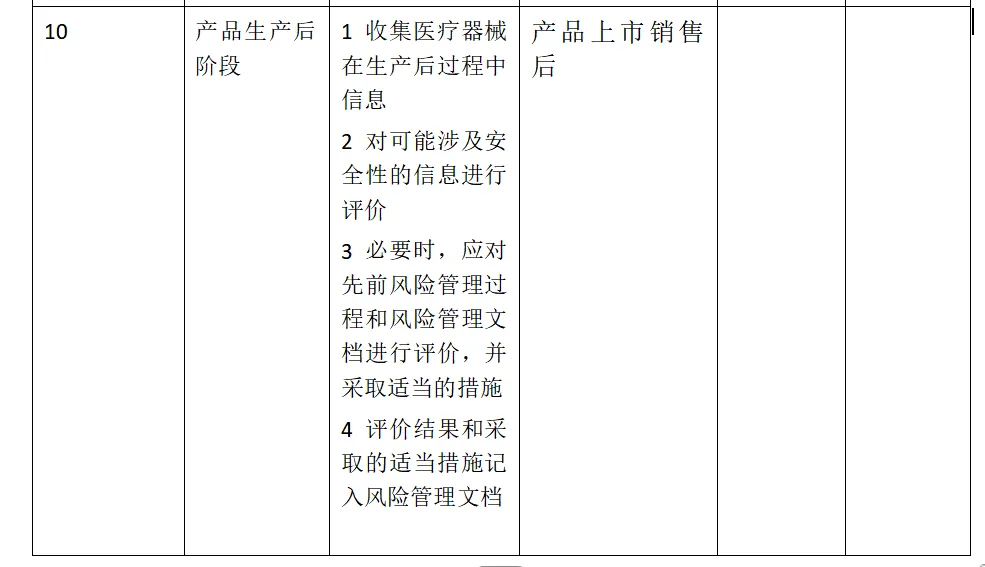
1.8风险管理活动评审的要求
评审组成员应对评审结果的正确性和有效性负责。
各部门应配合评审组成员利用《质量信息反馈控制程序》对与产品安全性有关的信息进行评审,为综合剩余风险的评价提供依据。
依据以下和安全性有关的信息在产品生命周期的各阶段进行评审:
1)是否有事先未知的危险出现;
2)是否有某项危险造成的已被估计的风险(一个或多个)不再是可接受的;
3)是否初始评定的其它方面已经失效;
4)产品综合剩余风险是否已降低至可接受水平或经过风险/受益分析判断为可接受。
应对产品生产和生产后信息的获取方式进行评审
保持评审记录以证实风险管理计划的每个要素在产品特定的生命周期阶段已被适当的实施。
评审组成员及其职责如下:
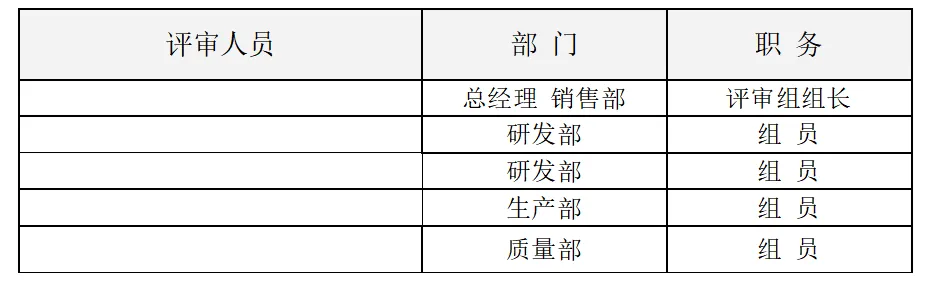
1.9风险管理活动验证的要求
1.9.1风险管理计划是否已适当实施的验证
评审组成员负责对风险管理计划的实施情况进行验证,以查看风险管理文档的方式查看风险分析、风险评价、风险控制等记录,确保风险管理计划策划的风险管理活动已得到适当的实施。
1.9.2风险管理活动效果的验证
评审组可通过收集临床资料及生产和生产后信息对风险管理实施效果进行验证以确保风险管理活动的有效性。
1.10生产和生产后信息收集
1.10.1 生产和生产后信息的收集情况,内容包括:
1)产品上市时间、上市数量、市场分布、使用客户群等
2.)制造依据、执行法规、质量体系情况
3)上市产品使用总体情况的概述
1.10.2生产和生产后信息的收集情况 包括:
a)生产过程和安全性有关的信息
b)来自安装和维修过程与安全性有关的信息
c)临床跟踪,包括
·本公司产品的不良事件情况
·来自顾客的与安全性相关的信息
·来自国内外医疗器械监管机构的同类产品不良事件报道
·来自国内外专业杂志有关同类医疗器械不良反应或不良事件的报告
d)新的或修订的法规和标准信息
1.10.3生产和生产后信息的评价
1)如有可能涉及安全性的信息,则列表说明
2)对设计安全性的信息进行评价
3)是否是先前没有认识的危险出现,如有,分项描述
4)是否由危险处境产生的一个或多个估计风险不再是可接受的
5)如3、4所述情况发生,则对先前风险管理活动的影响进行评价。
1.10.4 对先前的风险管理文档进行评审、对新的或不可接受的风险制定和实施风险控制措施,并对综合剩余风险和是否产生新的风险进行评价
如果出现一个或多个新的或不可接受的剩余风险,应对先前的风险管理文档进行评审,如先前的风险控制措施是否充分、得当等。
对新的或不可接受的风险制定和实施风险控制措施,并对综合剩余风险和是否产生新的风险进行评估
2、风险分析记录
1.1概述
本次风险分析就是对该产品从生物危险、能量危险、操作危险、信息危险等方面进行已知的和可预见的危险时间序列的一种危险分析,另外对产品在生命周期的各个阶段进行分析,包括危险分析和风险控制方案分析。
1.2风险分析人员
按照风险管理计划的安排,此次参加风险分析的部门包括生产部、质量部、研发部、销售部等。研发部主要分析设计开发阶段已知和可预见的危险时间序列,生产部和质量部主要分析产品生产阶段和已知和可预见的危险事件序列,销售部主要分析产品生产后已知和可预见的危险事件序列。研发部负责收集各部门分析的结果并按照法规和YY/T0316-2016附录E.1的资料对已知和可预见的危险序列进行分类,组织各部门进行风险评价和风险管理措施的分析与实施并编制相应的表格
1.3医疗器械预期用途和与安全有关特征的识别
安全特征问题清单及可能的危险,该清单依据 YY/T0316-2016标准附录C 的问题清单和附录E.1危险示例。
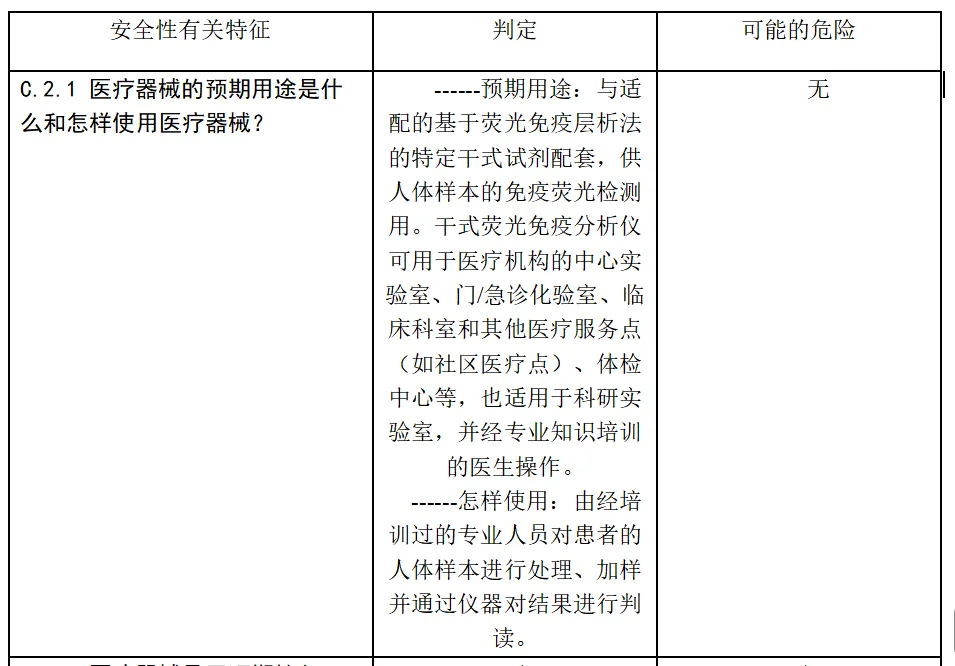
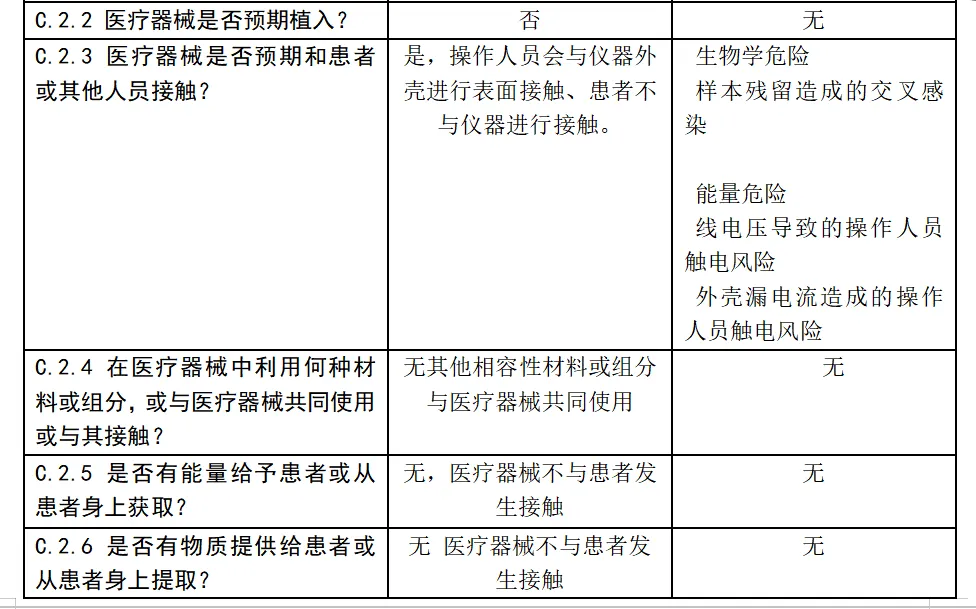
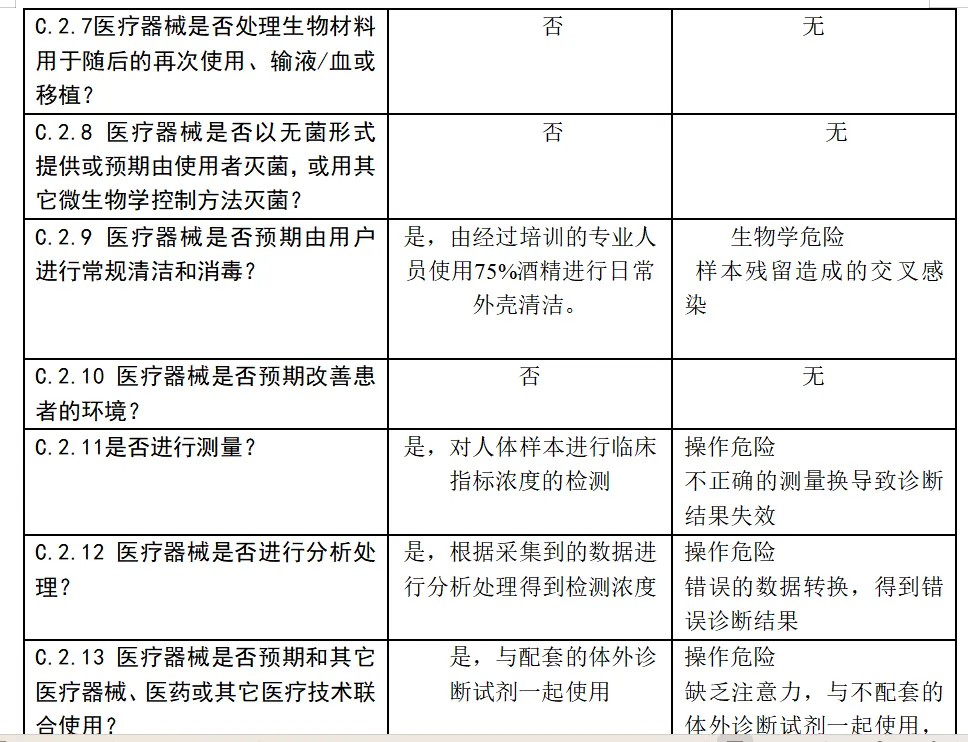
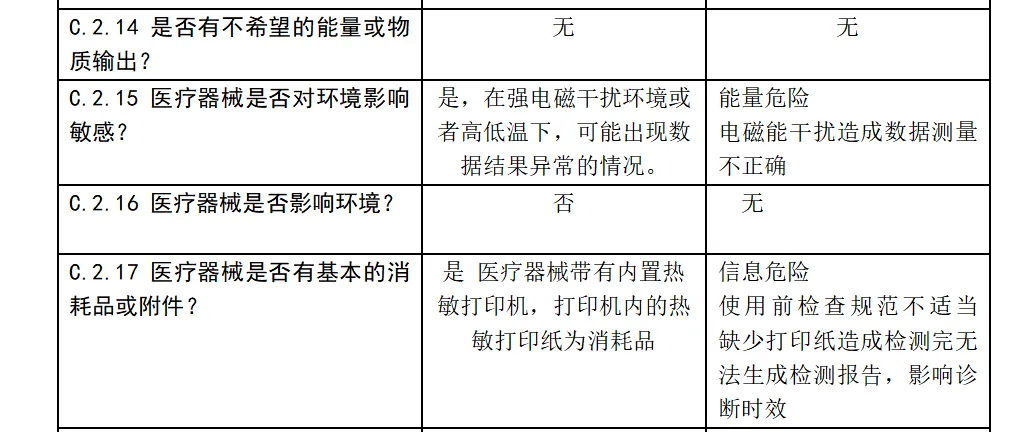
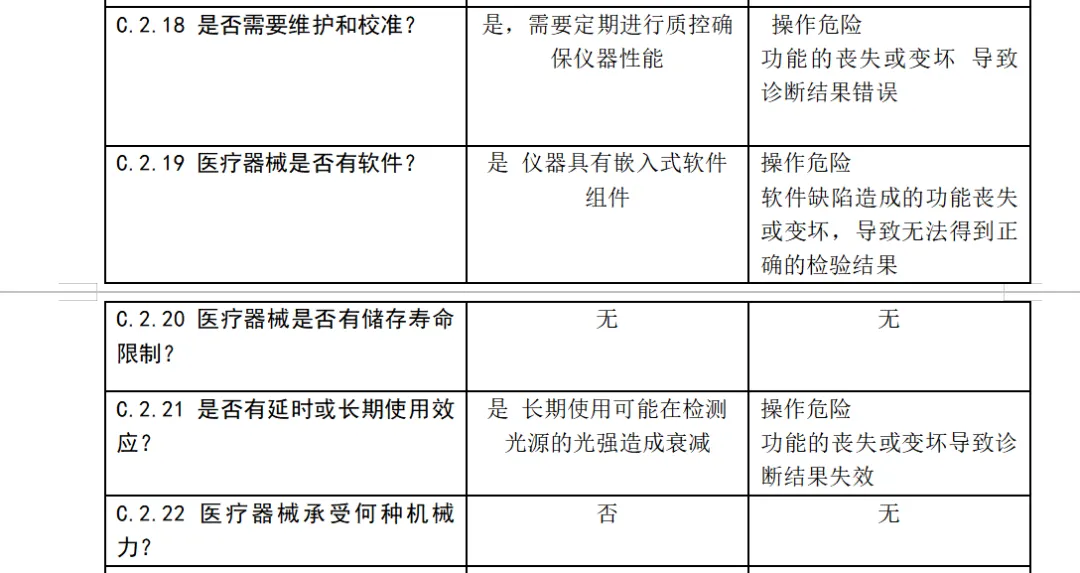
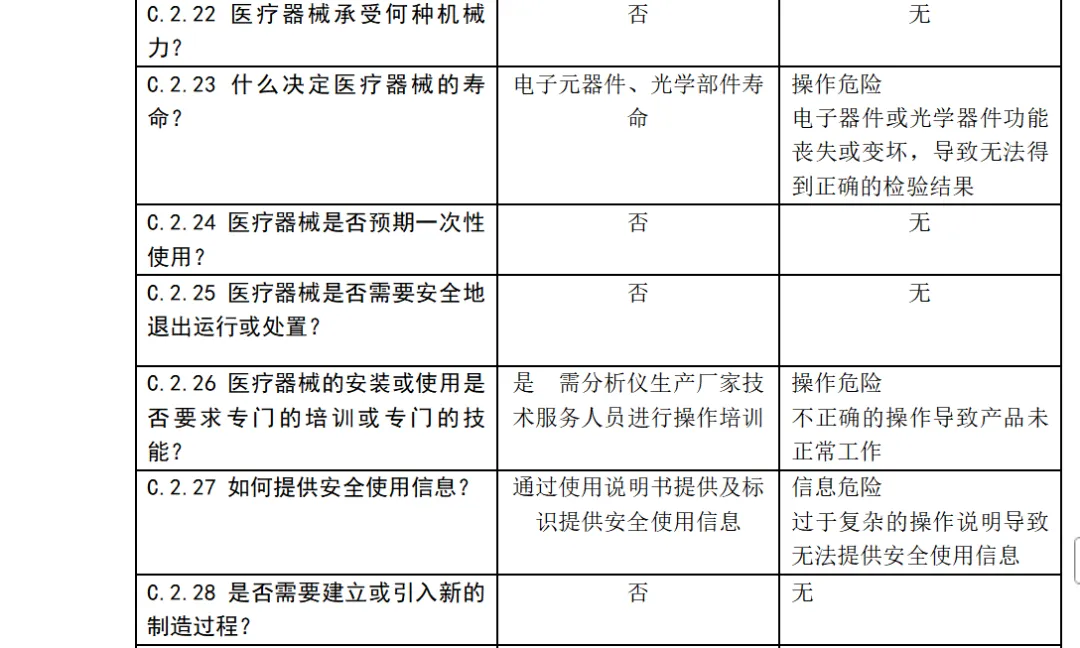
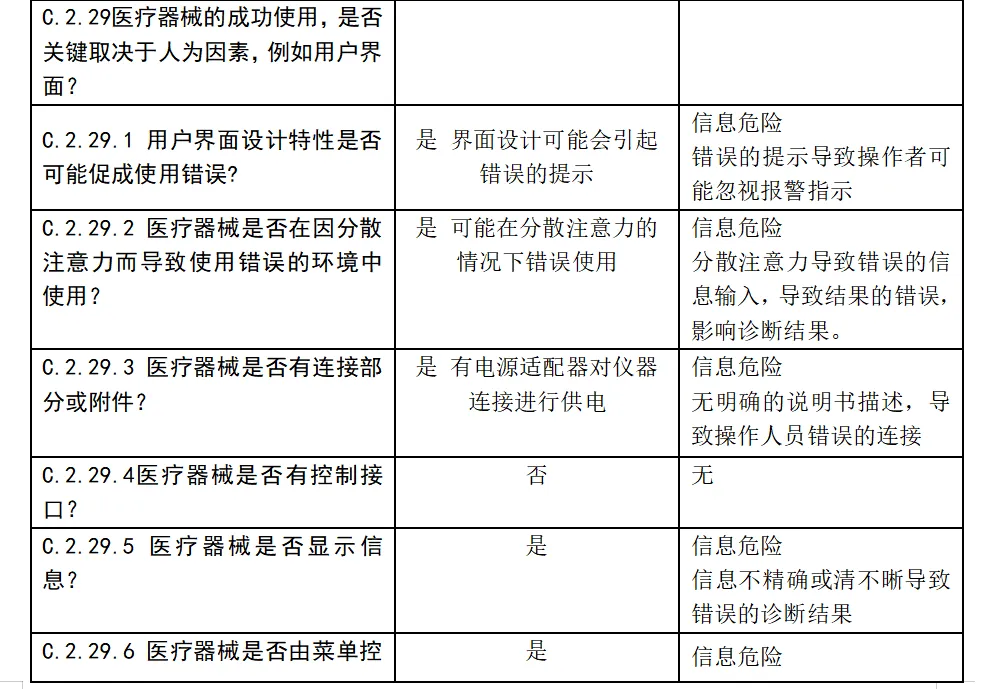
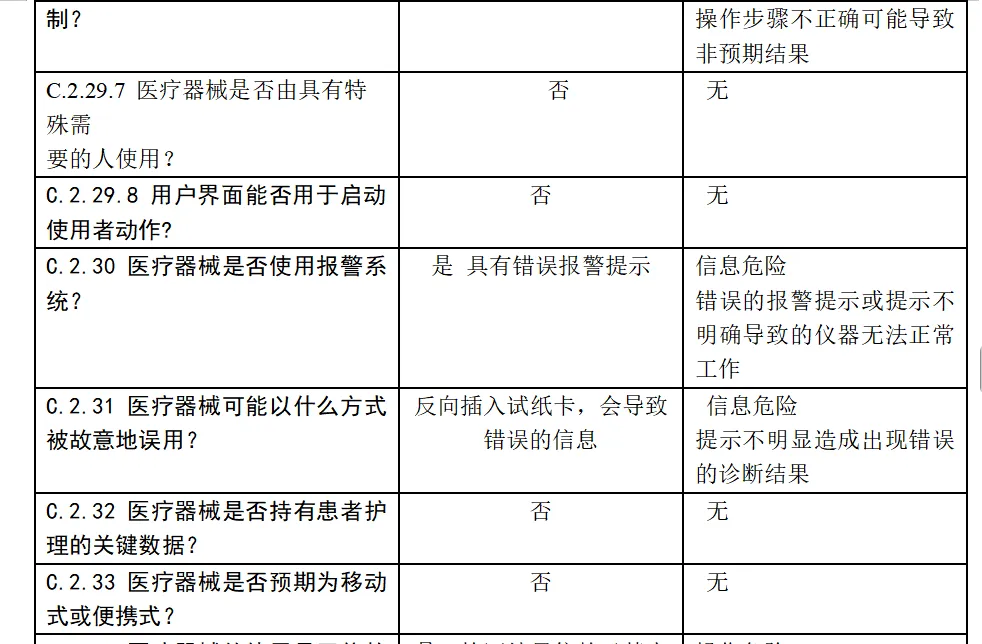

体外诊断医疗器械与安全性有关的特征判定
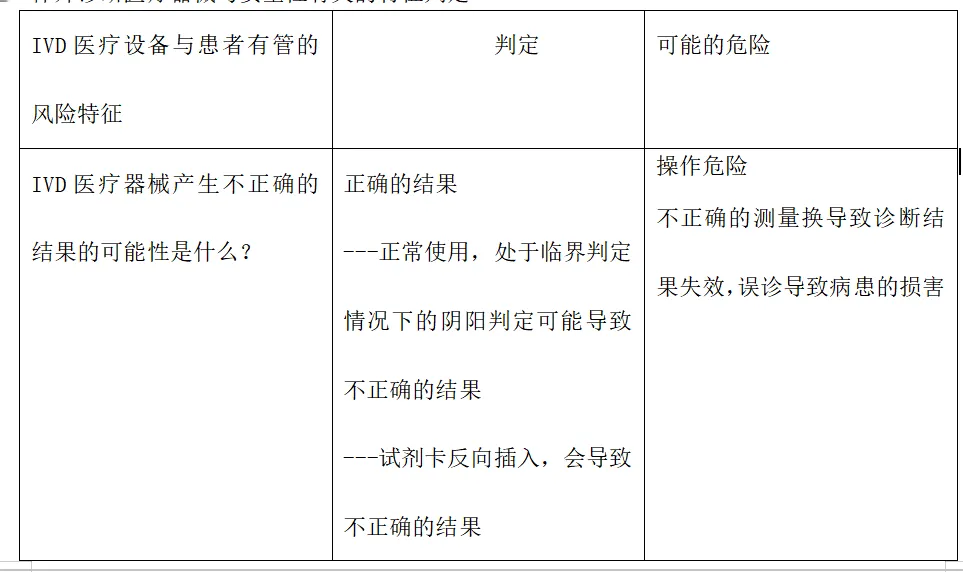
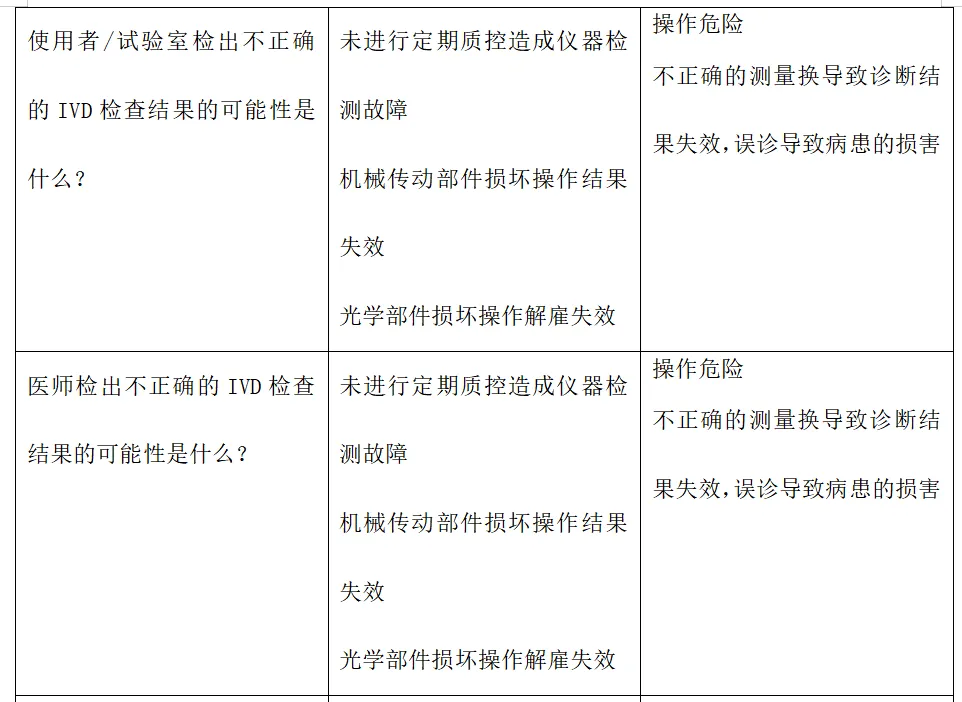
风险分析人员依据上表的提示,正常和故障状态下已知的和可预见的危险时间序列参考YY/T0316:2016危险示例进行分类,同时对可能发生的损害和初步控制措施进行分析,记录如下
分析分析过程包括了产品生命周期的全过程(包括产品设计、转换、更改、设计开发文档、合同评审、采购、供方控制、生产、检验、储存、交付、运输、售后服务等)
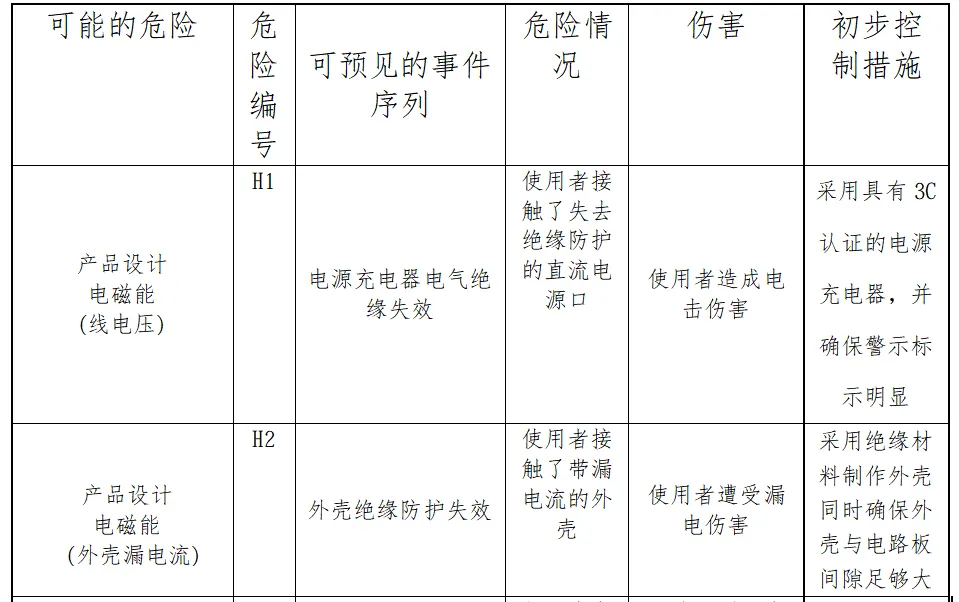
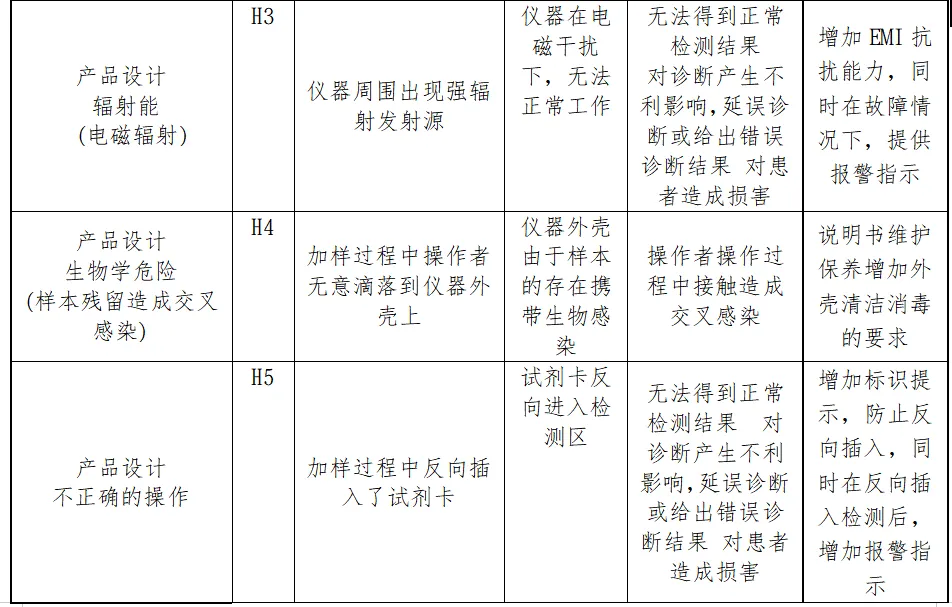
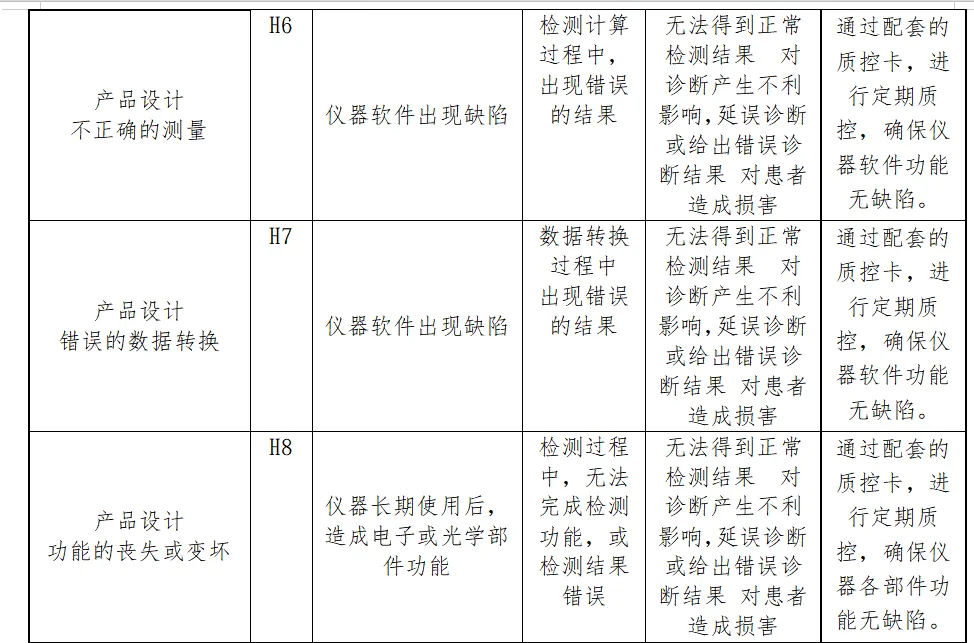
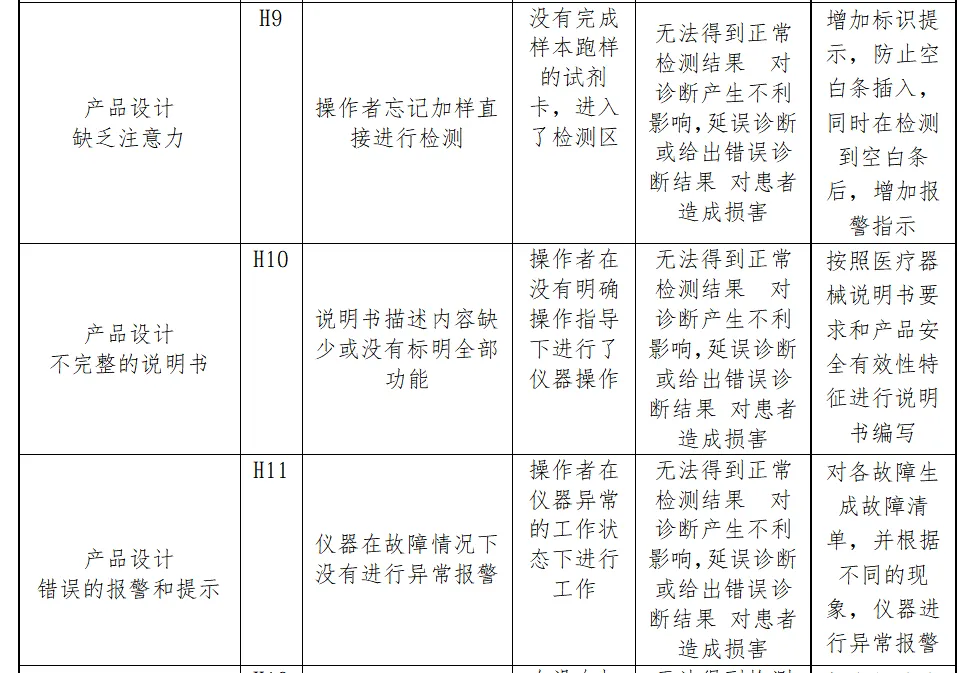
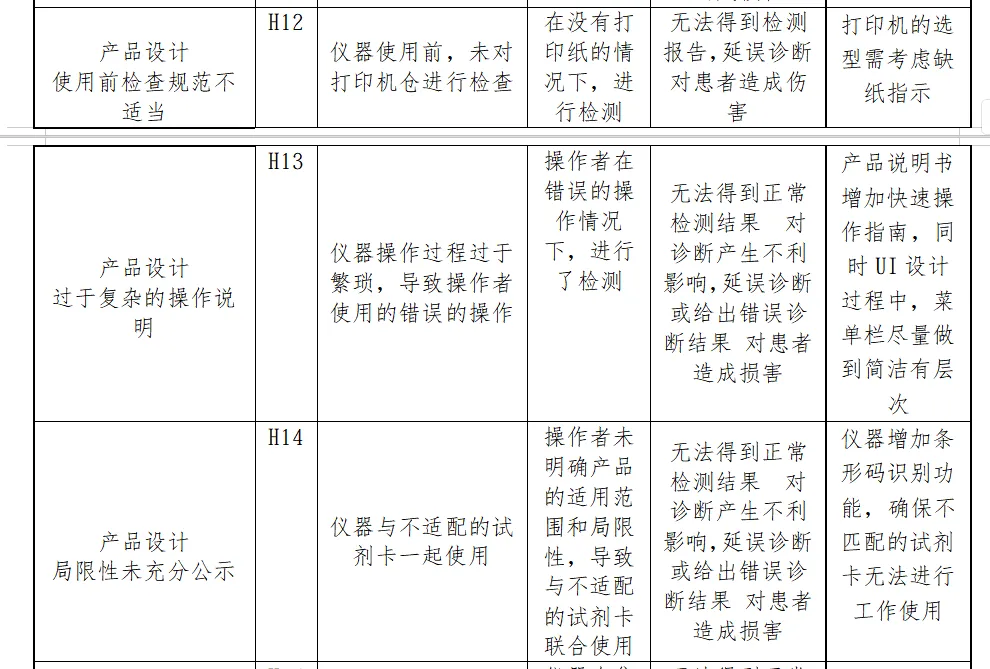
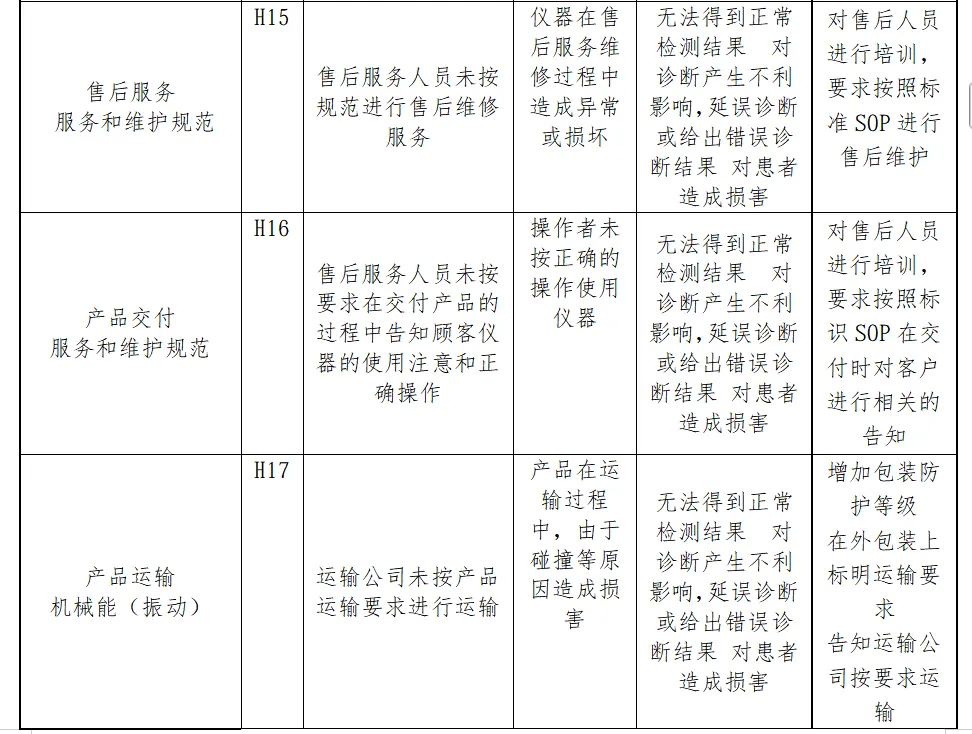
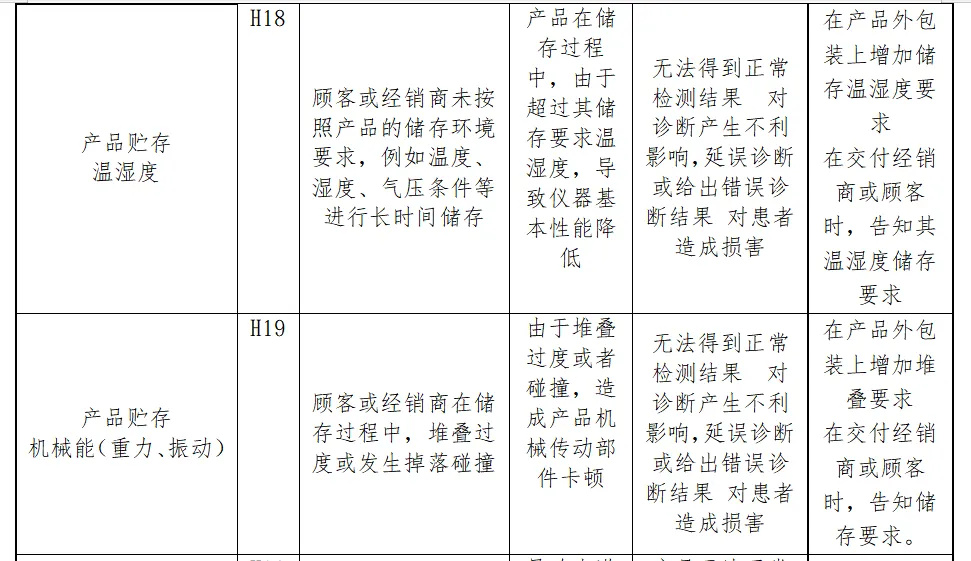
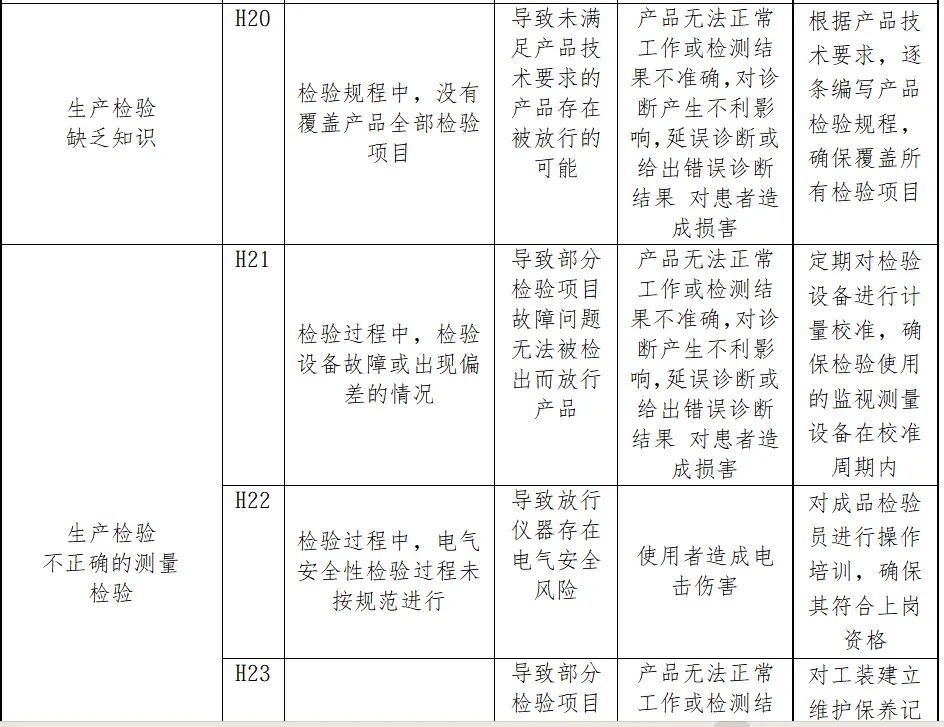
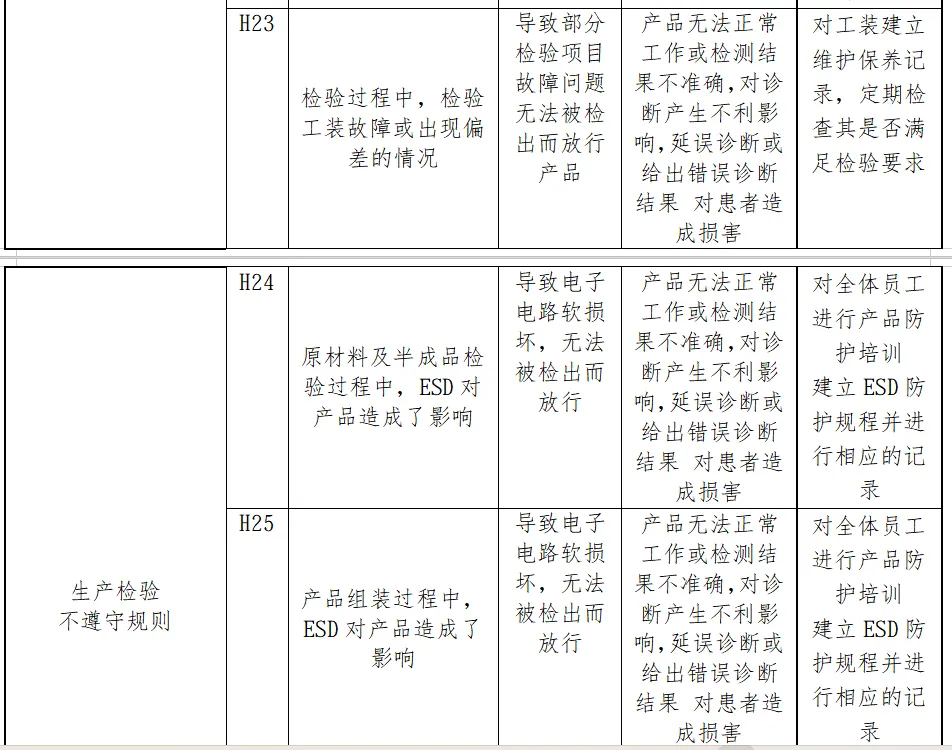
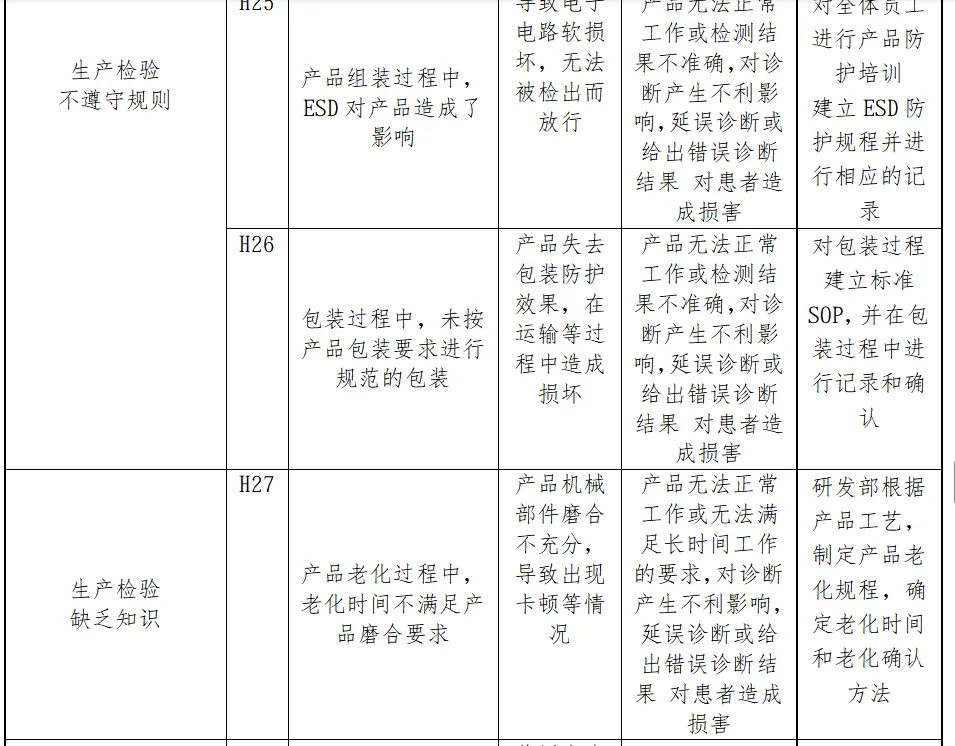
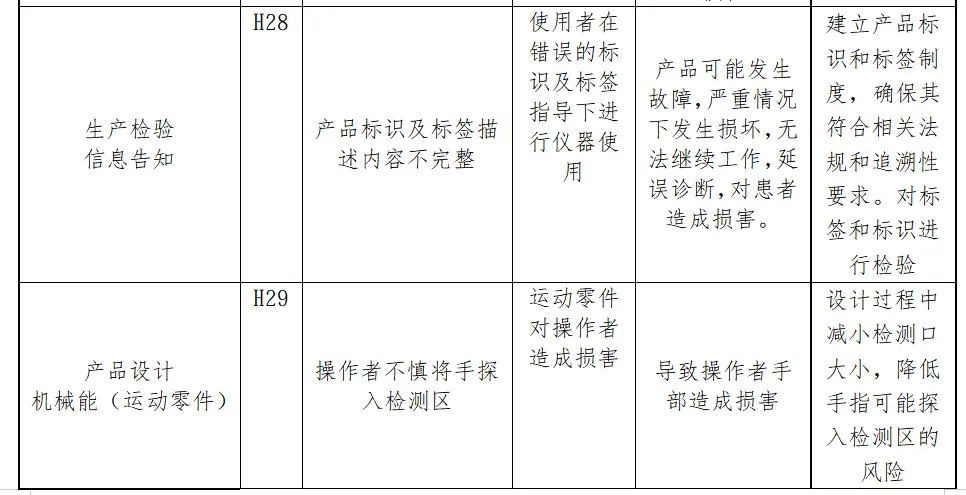
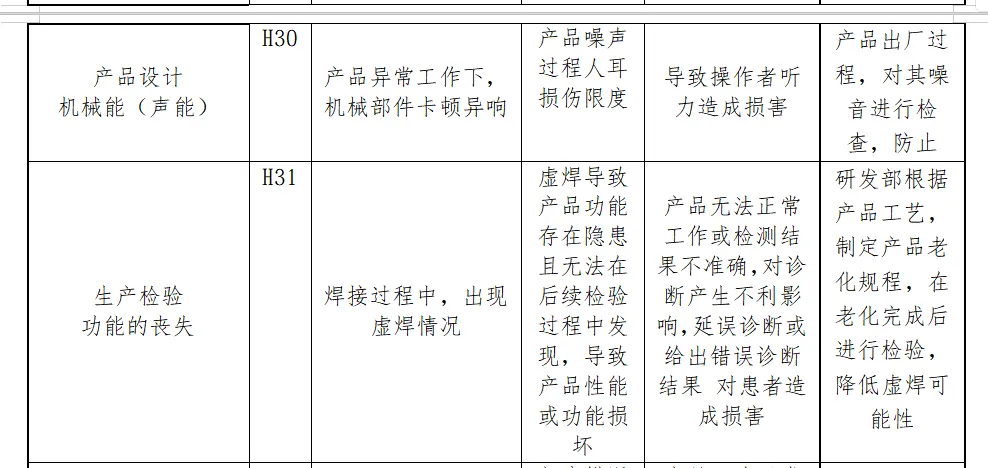
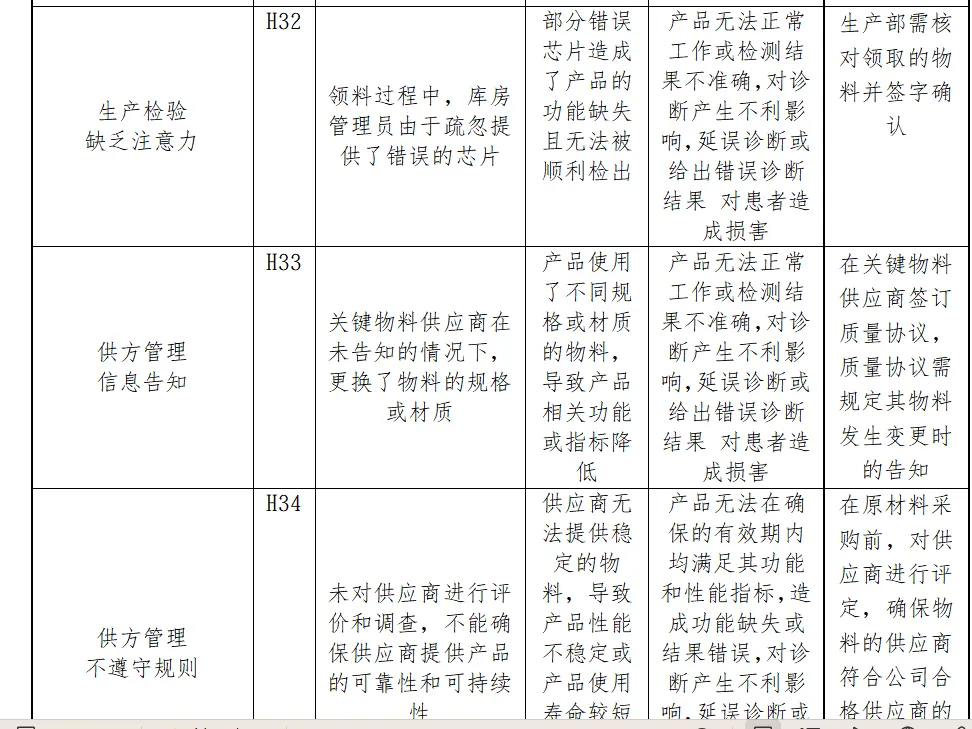
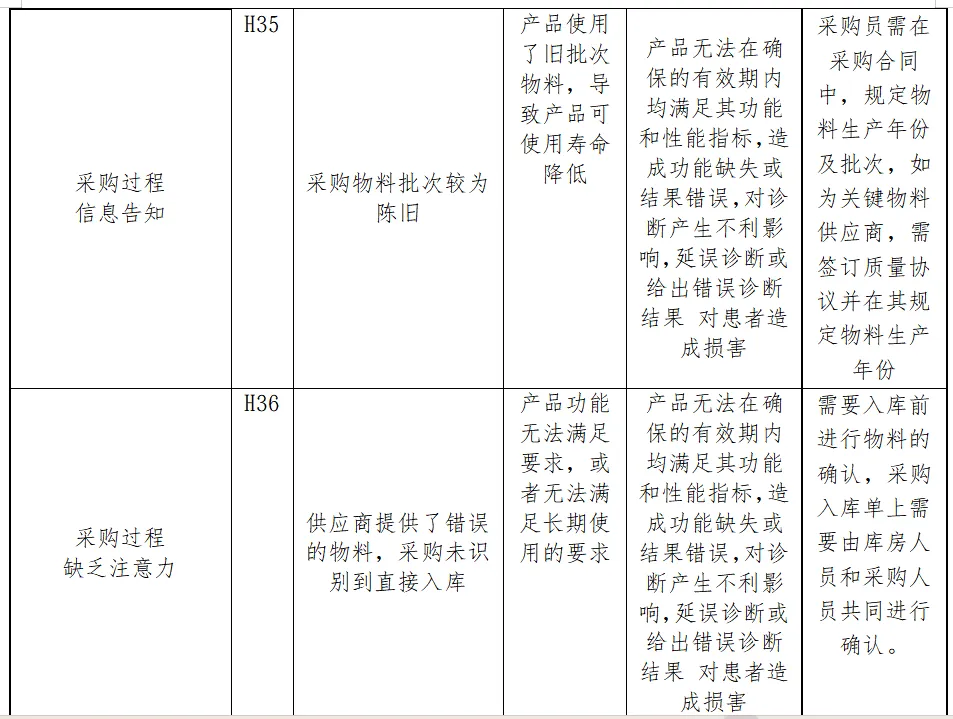
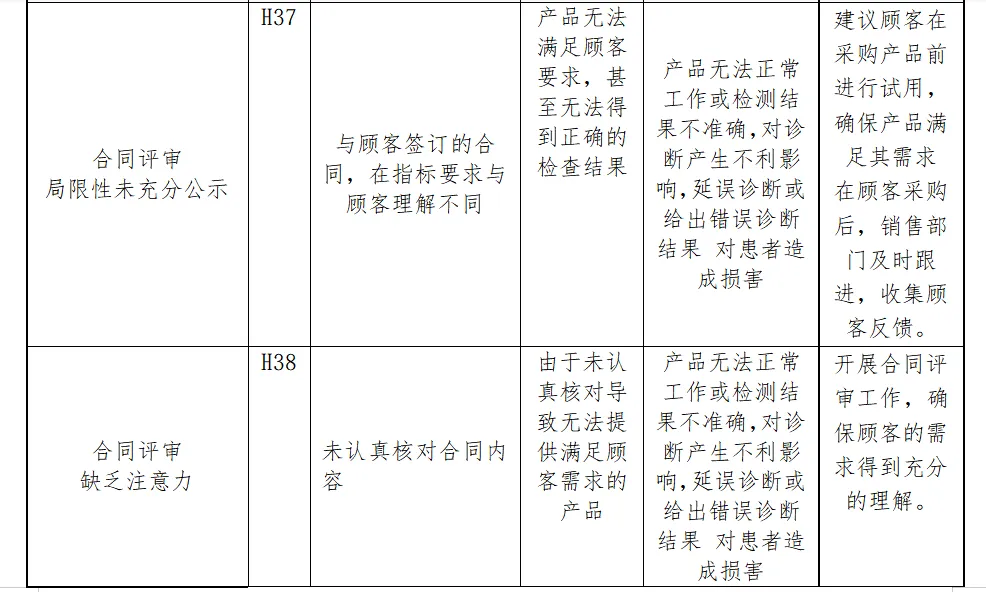
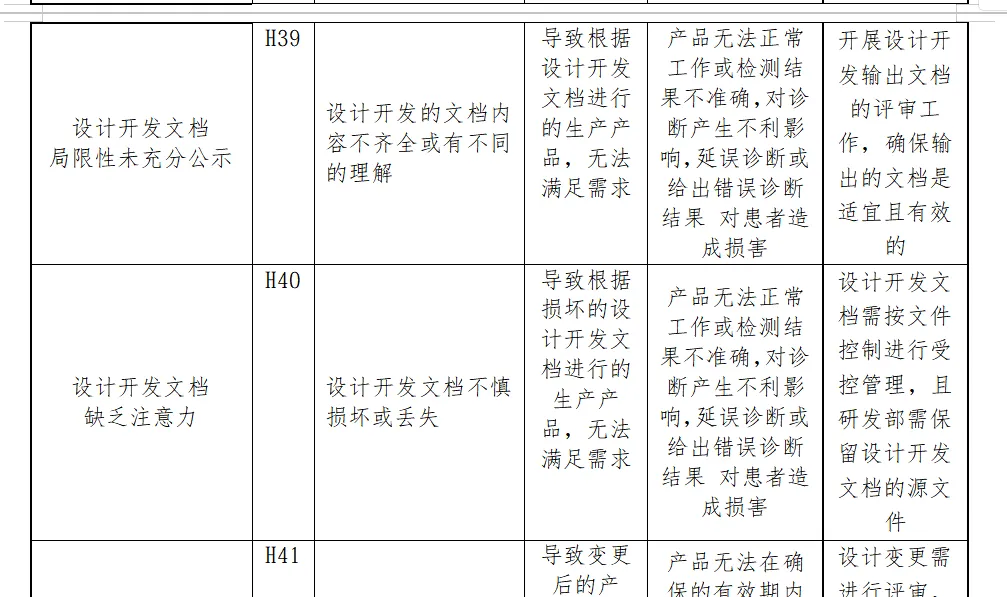
内容太多了,我就不一一展示了。
声明:这是一位工程师的辛苦劳作,感谢老师的创作,如有侵权请联系我18701083357。