 夜雨聆风
夜雨聆风





scDRS 实战教程:按官方文档跑通单细胞联合GWAS 分析
如果你前面看过一些 scDRS 介绍,却还是不知道“代码到底怎么跑”,这版就不再讲空泛概念,直接按 scDRS 官方仓库与文档 的结构来写:
-
项目仓库:https://github.com/martinjzhang/scDRS -
官方 CLI 文档:https://martinjzhang.github.io/scDRS/reference_cli.html -
MAGMA 转基因集说明:仓库 docs/compute_magma_gs.md
这篇文章重点回答 4 个问题:
-
环境怎么装; -
GWAS 结果怎么整理成 scDRS 能吃的 .gs文件; compute-score
和 perform-downstream怎么跑;-
输出文件怎么看,图该怎么配。
1. scDRS 到底在算什么
scDRS(single-cell Disease Relevance Score)用于把 GWAS 的疾病/性状信号 映射到 单细胞表达数据 上,最后回答的问题通常是:
-
哪些细胞类型与某个疾病更相关? -
同一种细胞内部,哪些细胞亚群更“疾病相关”? -
哪些连续变量(如 activation gradient、pseudotime、module score)和疾病相关性一起变化? -
哪些基因与 scDRS disease score 呈正相关?
如果你只记流程,可以先记这一条主线:
GWAS summary statistics → MAGMA gene statistics → scDRS
.gs→ 单细胞.h5ad→ compute-score → perform-downstream
2. 先准备好这几样输入
真正跑 scDRS 之前,先确认你手里至少有下面 3 类文件:
2.1 单细胞表达对象
官方 CLI 直接吃 .h5ad 文件,所以如果你的数据还在 Seurat 对象里,建议先转成 AnnData。
最低要求:
-
行/列方向正确; adata.var_names
是基因名; adata.obs
里至少有一个细胞分组字段,例如 cell_type;-
如果后面要做 group 分析 / corr 分析,对应列名要提前存在于 adata.obs。
2.2 GWAS 对应的基因层面统计
scDRS 自己不直接从原始 GWAS summary statistics 变出最终结果,推荐做法是先用 MAGMA 把 SNP 层结果聚合到基因层面,再转成 scDRS 的 .gs 文件。
2.3 可选协变量文件 .cov
如果你要在 compute-score 阶段纳入协变量,可以准备一个 .cov 文件。没有也可以先跑通主流程。
3. 环境安装:先把 CLI 跑起来
官方仓库基于 Python / AnnData / Scanpy。最稳的方式是单独建环境:
bash conda create -n scdrs python=3.9 -y conda activate scdrs pip install scdrs
装完先不要急着正式分析,先验证命令是否可用:
bash scdrs --help scdrs compute-score --help scdrs perform-downstream --help
官方文档特别提到:CLI 中 连字符
-和下划线_可以混用。比如compute-score与compute_score都能识别,但实战里建议统一写成官方示例风格,减少排错成本。
4. 第一步:把 GWAS 结果整理成 .gs
这一段是很多人最容易卡住的地方。
scDRS 官方推荐思路不是直接把 summary statistics 扔进去,而是:
-
先用 MAGMA 生成基因层面的统计量; -
再用 scdrs munge-gs把基因统计整理成.gs文件。
4.1 官方 MAGMA 流程里最关键的一步
在仓库 docs/compute_magma_gs.md 里,官方给出的核心 MAGMA 计算命令是:
bash mkdir -p out/step2 ${magma_dir}/magma \ --bfile ${magma_dir}/g1000_eur \ --pval ${trait}.pval use='SNP,P' ncol='N' \ --gene-annot out/step1.genes.annot \ --out out/step2/${trait}
这里你至少要注意 3 件事:
trait.pval
里要有 MAGMA 所需列; N
(样本量)不能乱填; out/step1.genes.annot
是前一步 annotate 产生的文件,不能缺。
4.2 把 MAGMA 结果转成 scDRS .gs
官方 CLI 的 munge-gs 用来把 基因层面 p 值或 z-score 表 转成 .gs 文件。
官方文档中的典型写法:
bash scdrs munge-gs \ --out-file disease.gs \ --zscore-file disease_zscore.tsv \ --weight zscore \ --n-max 1000
如果你要用 p 值文件,也可以走 --pval-file。
官方要求这类输入表的第一列是 GENE,后面每一列对应一个 trait,例如:
tsv GENE BMI HEIGHT OR4F5 0.001 0.01 DAZ3 0.01 0.001
实战建议:
-
先只放 1 个 trait 做测试; -
先保证 GENE列能和单细胞对象里的基因名体系对上; -
物种要提前统一,人/鼠混错是高频报错源。
5. 第二步:正式计算 scDRS score
当你已经有:
-
单细胞 adata.h5ad -
trait 基因集 disease.gs -
可选协变量 covariates.cov
就可以进入核心命令:compute-score。
5.1 官方 CLI 示例
下面这段基本就是官方文档里的标准骨架:
bash scdrs compute-score \ --h5ad-file data/example.h5ad \ --h5ad-species mouse \ --gs-file gwas/disease.gs \ --gs-species human \ --out-folder results/score \ --cov-file meta/covariates.cov \ --flag-filter-data True \ --flag-raw-count True \ --n-ctrl 1000 \ --flag-return-ctrl-raw-score False \ --flag-return-ctrl-norm-score True
5.2 这些参数最值得先看懂
--h5ad-file
你的单细胞表达对象。
--h5ad-species
单细胞对象的物种,可用值包括 human / hsapiens / mouse / mmusculus。
--gs-file
上一步整理好的 scDRS 基因集文件。
--gs-species
GWAS 基因集的物种。很多项目是 人类 GWAS + 小鼠单细胞,这时这个参数和 --h5ad-species 不一定相同。
--n-ctrl
控制基因集的数量。官方文档说明:后续 MC 检验的精度会受这里影响。一般先按 1000 起步。
--flag-filter-data
是否做最基本的 cell / gene filtering。
--flag-raw-count
如果输入还是 raw count,通常设为 True,让 scDRS 做 size-factor normalization 和 log1p 处理。
如果你的
.h5ad已经是处理过的表达矩阵,就不要机械照抄参数,先确认对象状态。
5.3 跑完会生成什么
官方说明里,compute-score 会为每个 trait 产出两类核心文件:
<trait>.score.gz<trait>.full_score.gz
可以把它们理解成:
score.gz
:常用汇总结果; full_score.gz
:包含后续下游分析要用的完整信息,做 downstream 时要重点用这个。
6. 第三步:做 group / corr / gene 下游分析
当 full_score.gz 已经跑出来之后,就可以继续 perform-downstream。
6.1 官方示例命令
bash scdrs perform-downstream \ --h5ad-file data/example.h5ad \ --score-file results/score/IBD.full_score.gz \ --out-folder results/downstream \ --group-analysis cell_type \ --corr-analysis causal_variable,non_causal_variable,covariate \ --gene-analysis \ --flag-filter-data True \ --flag-raw-count True
6.2 这条命令在做什么
--group-analysis cell_type
按 adata.obs['cell_type'] 做细胞群层面的疾病关联分析。
输出里你会得到每种细胞类型与 trait 的关联强弱,以及群内异质性信息。
--corr-analysis ...
对单细胞层面的连续变量做相关性检验。
比如你可以换成自己的字段:
bash --corr-analysis pseudotime,module_score,activation_score
前提是这些列确实存在于 adata.obs。
--gene-analysis
计算每个基因表达与 scDRS disease score 的相关性,方便后续找 marker / driver-like signal。
7. 输出文件怎么看:别只盯着图
官方 file_format.rst 对输出解释得很清楚,这里提炼成最实用版本。
7.1 <trait>.scdrs_group.<annot>
这个文件对应 细胞群层面分析。
你至少要看这些列:
n_cell
:该组细胞数 assoc_mcp
:群体与疾病关联的 MC p 值 assoc_mcz
:群体与疾病关联的 MC z-score hetero_mcp
:群内异质性 MC p 值 hetero_mcz
:群内异质性 MC z-score
最简单的解释方式是:
assoc_mcp
越小,说明这个细胞群整体越值得关注; assoc_mcz
越高,通常说明富集信号越强; -
如果 hetero_*也明显,说明同一种细胞内部不是“所有细胞一样相关”。
7.2 <trait>.scdrs_cell_corr
这个文件对应 单细胞变量相关性分析。
核心列:
corr_mcpcorr_mcz
如果某个变量在这里显著,说明它和 scDRS disease score 有同步变化趋势。
7.3 <trait>.scdrs_gene
这个文件对应 基因层面的相关性结果。
关键列:
CORRRANK
最常见用法是:
-
取 CORR高的基因做 marker 回看; -
结合已知通路或免疫/代谢/应激基因集解释细胞状态。
8. 文章里怎么配图:直接参考官方仓库风格
你前面提到“没有图片”,这个意见是对的。scDRS 这类教程如果只有文字,很难让读者形成结果直觉。
官方仓库 README 里已经给了很适合做文章配图的示意图,最直接的是下面两张:
图 1:TMS FACS 全局细胞图谱
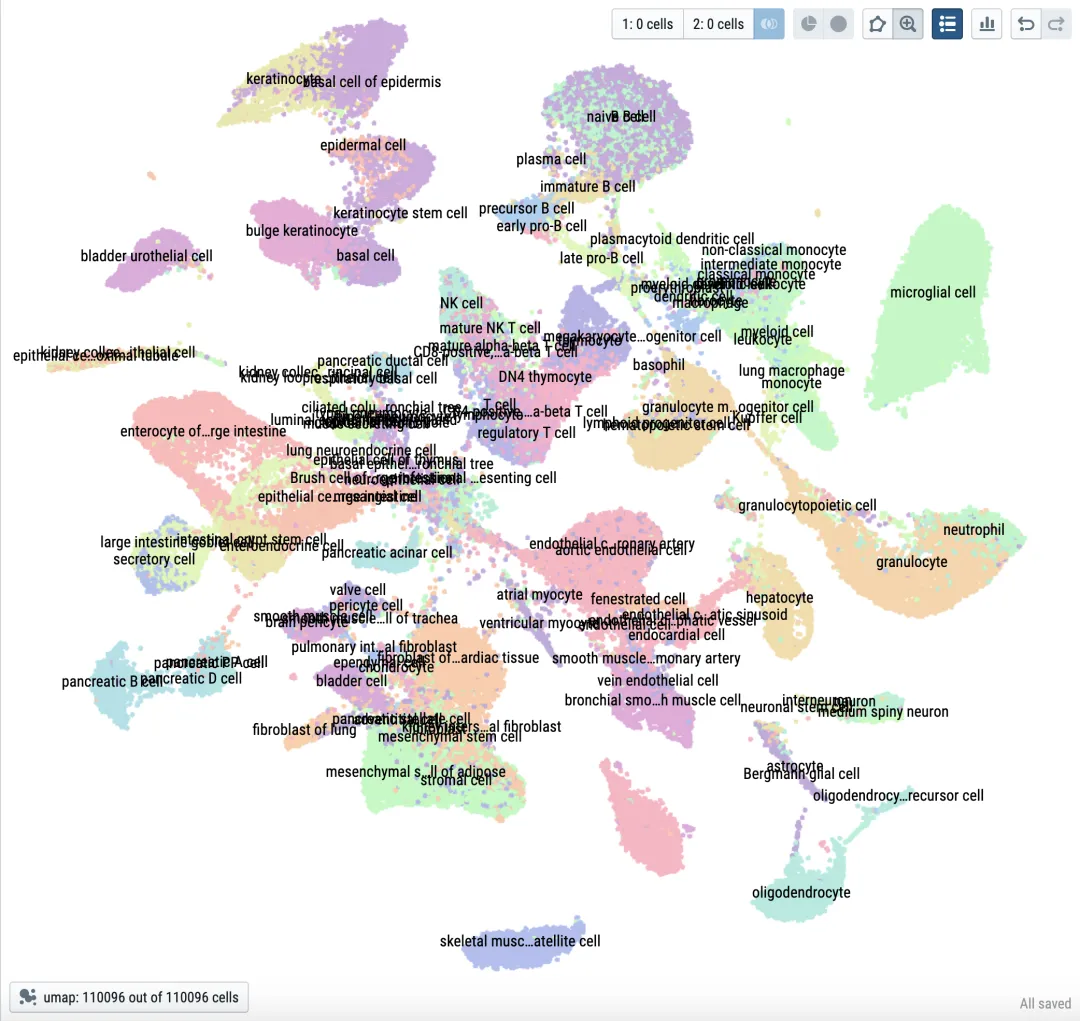
scDRS 官方示意图:TMS FACS 全局细胞图谱
建议配图说明:
这是官方 README 中用于展示 scDRS 结果浏览方式的整体细胞图谱。写文章时,这张图适合放在“scDRS 能看到什么”这一节,告诉读者结果最终不是一个抽象分数,而是能落到具体细胞空间中的。
图 2:IBD-associated cells 示例
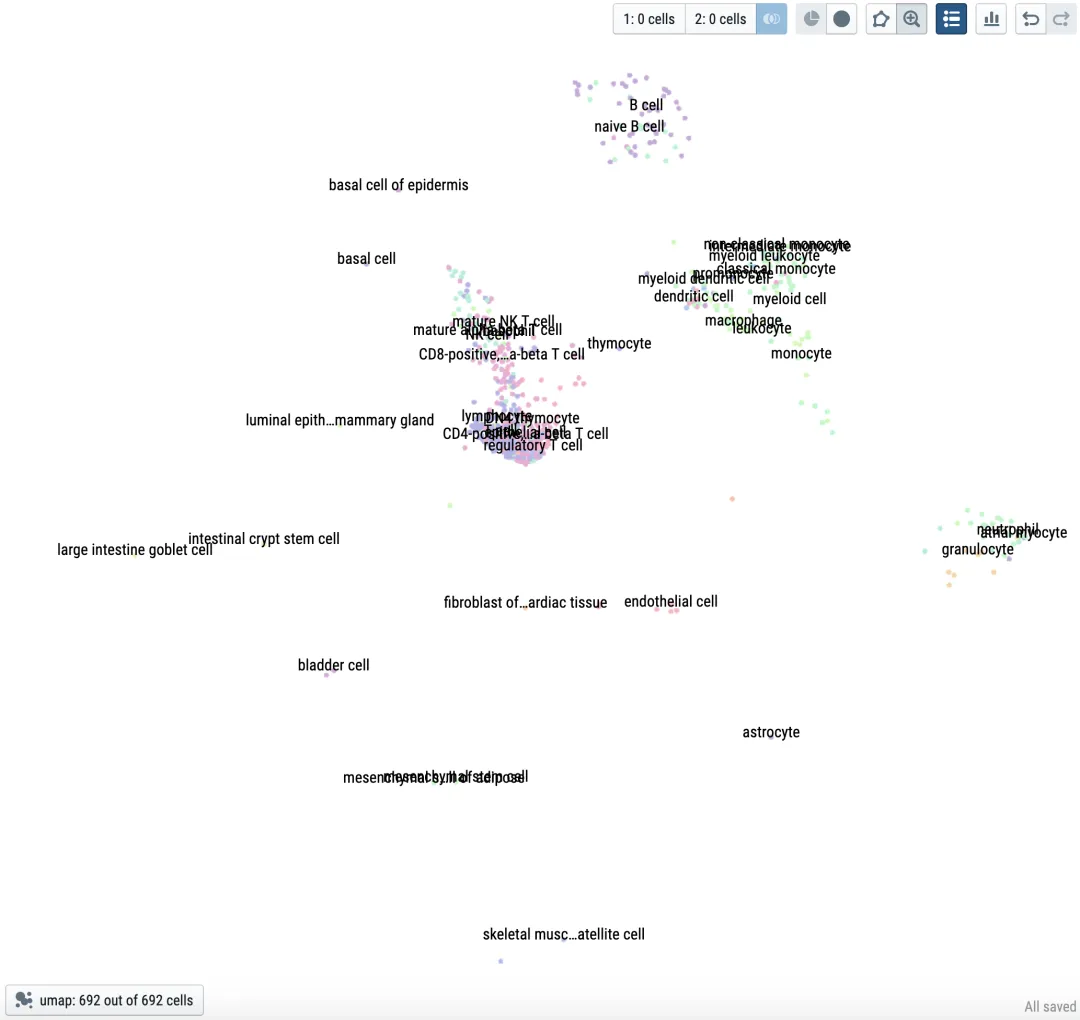
scDRS 官方示意图:IBD-associated cells
建议配图说明:
这张图更适合放在“下游结果解读”部分。它直观展示了某个 trait(这里是 IBD)相关细胞如何在嵌入空间里聚集,比单纯贴一张表更有说服力。
如果你后续自己出图,建议最少准备这 3 类:
group-analysis
的细胞类型排名图; -
某个显著细胞群的 UMAP / tSNE 着色图; gene-analysis
里 top genes 的热图或条形图。
9. 一个最小可复现命令清单
如果你只想先跑通一次,不想一上来就做完整项目,可以按下面这个顺序:
Step 1:安装
bash conda create -n scdrs python=3.9 -y conda activate scdrs pip install scdrs
Step 2:准备基因集
bash scdrs munge-gs \ --out-file ibd.gs \ --zscore-file ibd_magma_zscore.tsv \ --weight zscore \ --n-max 1000
Step 3:算 disease score
bash scdrs compute-score \ --h5ad-file data/pbmc.h5ad \ --h5ad-species human \ --gs-file ibd.gs \ --gs-species human \ --out-folder out/score \ --flag-filter-data True \ --flag-raw-count True \ --n-ctrl 1000
Step 4:做细胞类型分析
bash scdrs perform-downstream \ --h5ad-file data/pbmc.h5ad \ --score-file out/score/IBD.full_score.gz \ --out-folder out/downstream \ --group-analysis cell_type \ --gene-analysis \ --flag-filter-data True \ --flag-raw-count True
Step 5:先检查这 3 份结果
out/score/IBD.score.gzout/downstream/IBD.scdrs_group.cell_typeout/downstream/IBD.scdrs_gene
如果这 3 份都能正常生成,你的主流程基本就通了。
10. 高概率踩坑点
10.1 物种不一致
这是最常见的问题之一。
-
GWAS gene set 是 human -
单细胞对象是 mouse
这种场景不是不能做,但你必须明确自己的跨物种映射策略,不能直接忽略。
10.2 基因名体系对不上
比如一边是 Ensembl ID,一边是 gene symbol;或者同义名没有统一,都会导致实际参与计算的基因数明显下降。
10.3 .h5ad 不是你以为的 raw count
--flag-raw-count True 默认意味着 scDRS 要继续做 normalization + log1p。如果你的矩阵已经处理过,就要先核对,不要重复处理。
10.4 adata.obs 里根本没有你写的字段
像下面这种写法:
bash --group-analysis cell_type --corr-analysis pseudotime,module_score
要求 adata.obs 里真的有 cell_type、pseudotime、module_score 这些列,否则一定报错。
10.5 还没跑通就急着上大队列
最稳的策略永远是:
-
先拿一个小 .h5ad测试; -
先只放一个 trait; -
先把 compute-score跑通; -
再做 downstream; -
最后再扩展到完整数据集。
11. 这版文章和前一版有什么不同
如果说前一版更像“思路介绍”,那这版就明确改成 教程体 了,重点变化有 3 个:
- 加入官方 CLI 代码块
:不再只讲概念; - 加入图片
:直接引用 scDRS README 中的官方示意图; - 按可执行顺序组织
:安装 → 准备 .gs→compute-score→perform-downstream→ 看输出。
如果你后面要继续发公众号,这一版已经更接近“读者照着就能跑”的风格了。下一步最值得补的,是:
-
再加一个“Seurat 转 h5ad”的前处理小节; -
再加一段“真实结果截图 + 参数解释”; -
如果有你自己的数据结果,再把官方示意图替换成你自己的 UMAP / barplot。
12. 小结
做 scDRS,最重要的不是背命令,而是把这条链路打通:
GWAS → MAGMA gene statistics → scDRS
.gs→.h5ad→compute-score→perform-downstream→ 结果解释
最后再补一个更实用的内容:我这边已经基于生信服务器搭好了 scDRS 在线分析环境,并录制了对应的演示视频。如果你想知道这套流程在真实界面里是怎么操作的,可以直接看下面的视频。相比只看命令行,这种方式会更容易理解整个分析过程。