 夜雨聆风
夜雨聆风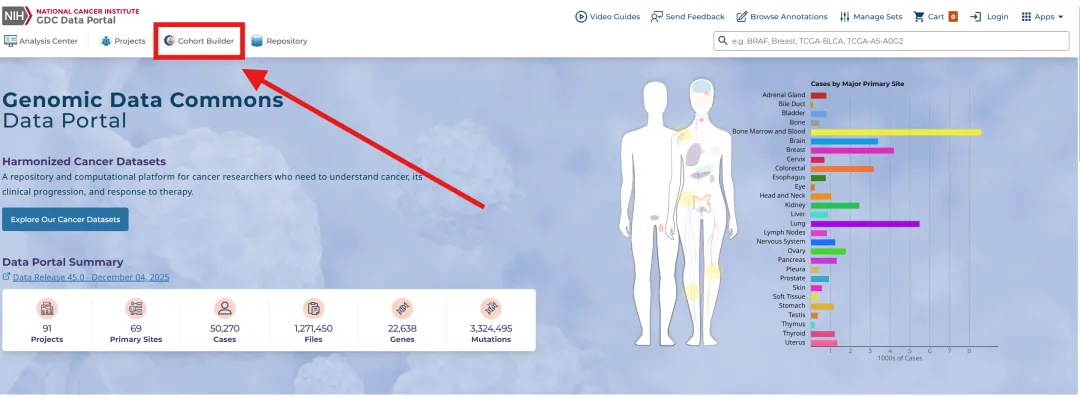
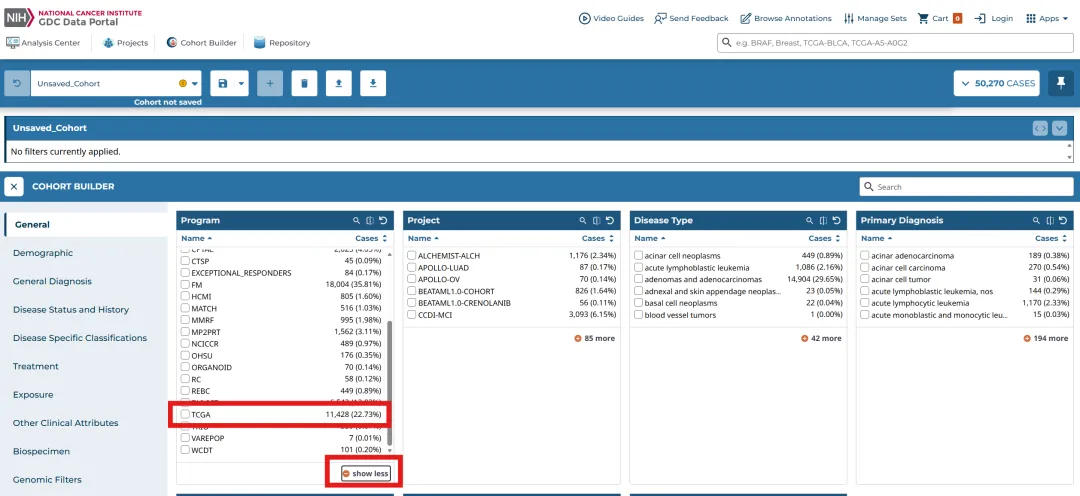
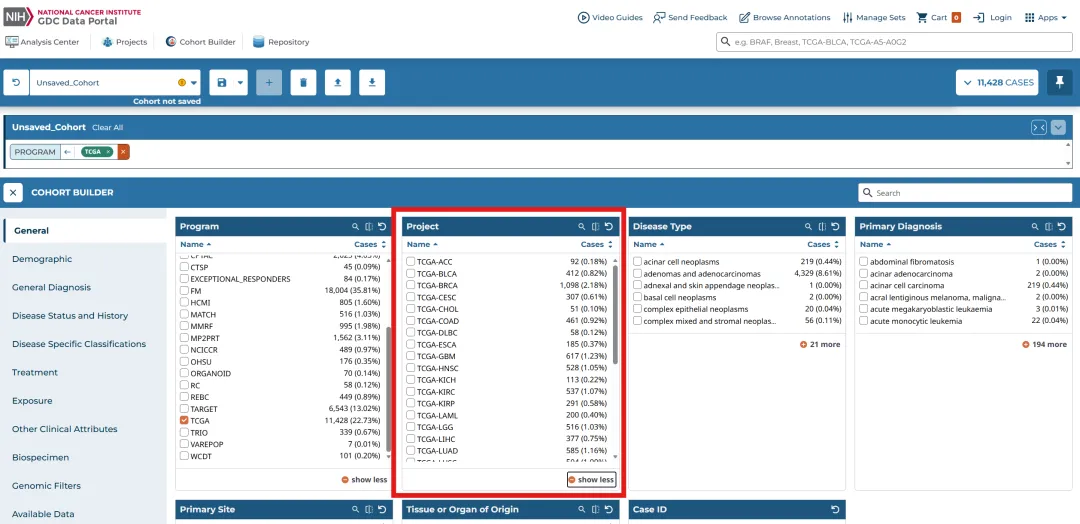
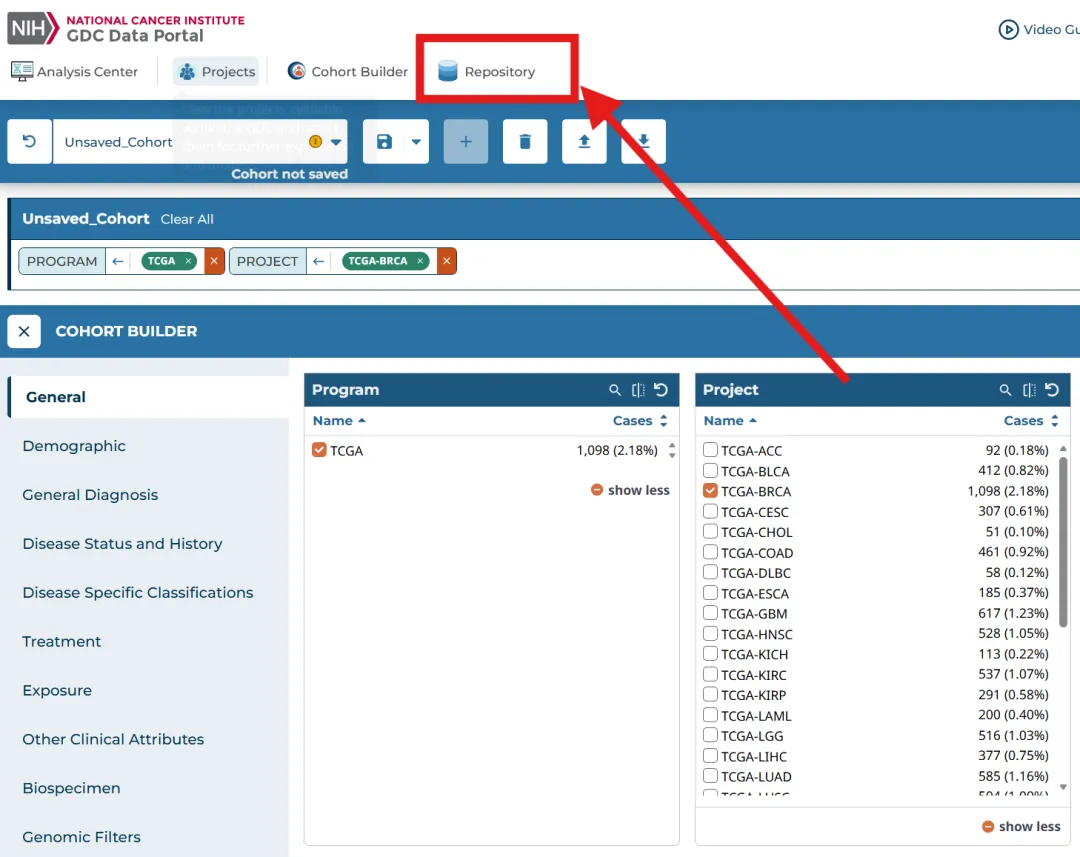
library(TCGAbiolinks)library(limma)library(future.apply)library(pbapply)library(parallel)library(doParallel)library(data.table)setDTthreads(threads =detectCores())plan(multisession)source("xxdTCGAmerge.R",encoding = "utf-8")
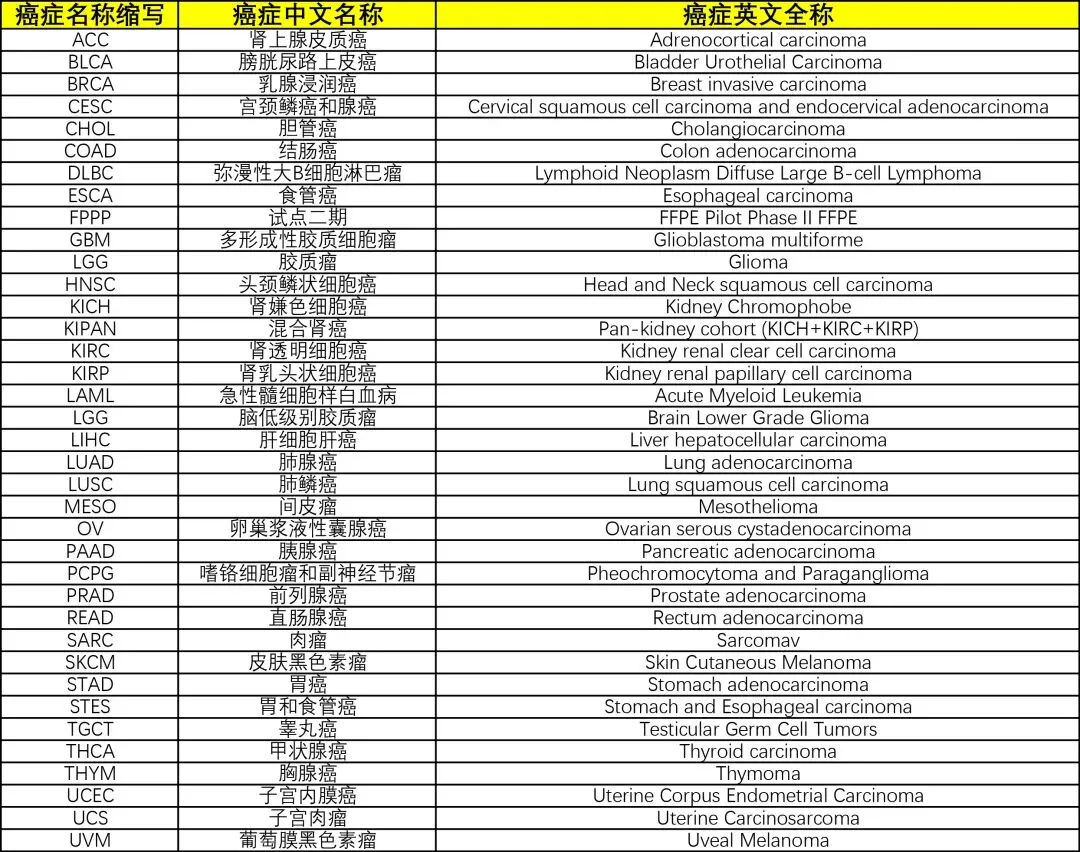
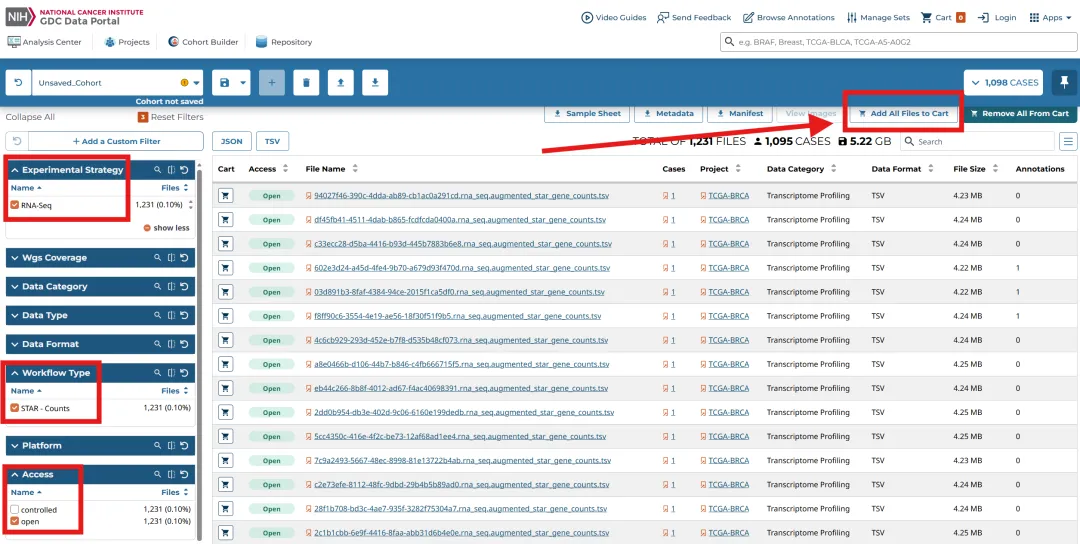
tissue<-"TCGA-BRCA"TCGAdata<-GDCquery(project = tissue,data.category = "Transcriptome Profiling",data.type = "Gene Expression Quantification")GDCdownload(TCGAdata)
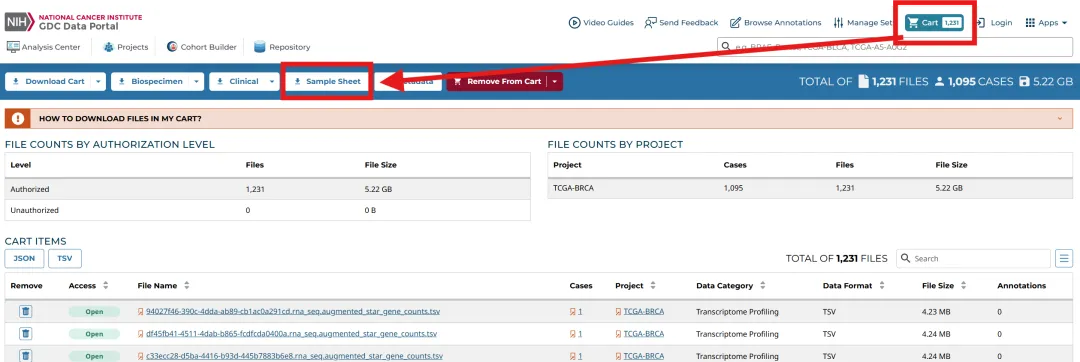
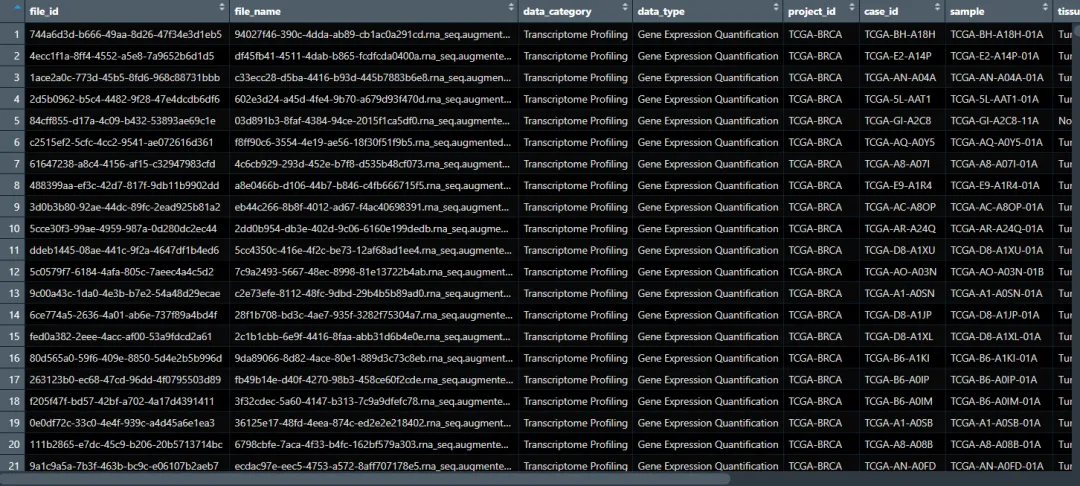
metaMatrix.RNA=read.table("gdc_sample_sheet.2026-03-06.tsv",sep="\t",header=T)names(metaMatrix.RNA)=gsub("sample_id","sample",gsub("\\.","_",tolower(names(metaMatrix.RNA))))metaMatrix.RNA$sample_type=gsub(" ","",metaMatrix.RNA$tumor_descriptor)metaSample<-list.files(path = paste0(".\\GDCdata\\",tissue,"\\Transcriptome_Profiling\\Gene_Expression_Quantification"))samemeta<-intersect(metaMatrix.RNA$file_id,metaSample)metaMatrix.RNA<-metaMatrix.RNA[metaMatrix.RNA$file_id %in% samemeta,]
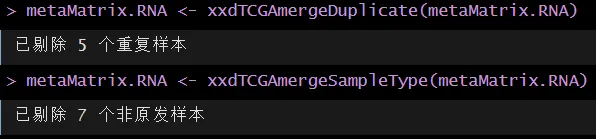
metaMatrix.RNA <- xxdTCGAmergeDuplicate(metaMatrix.RNA)metaMatrix.RNA <- xxdTCGAmergeSampleType(metaMatrix.RNA)
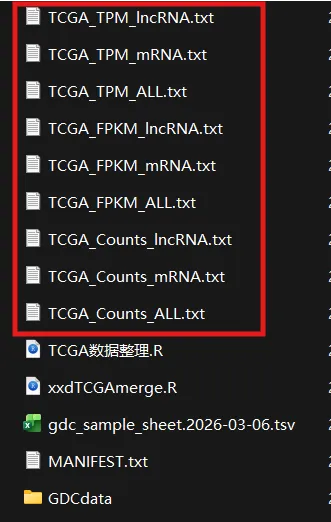
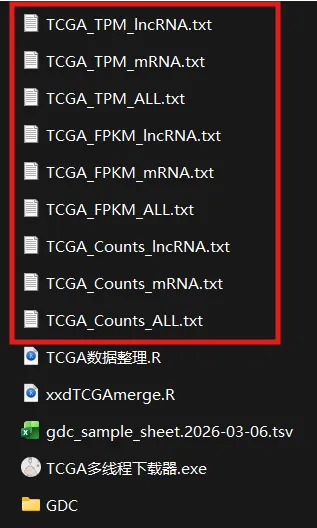
lapply(c("STAR","FPKM","TPM"),function(exptype){namestype<-ifelse(exptype=="STAR","Counts",ifelse(exptype=="FPKM","FPKM","TPM"))RNA_seq<-xxdTCGAmerge(metadata=metaMatrix.RNA,path=paste0(".\\GDCdata\\",tissue,"\\Transcriptome_Profiling\\Gene_Expression_Quantification"),data.type="RNAseq",mRNA_expr_type=exptype,symbol = T,RNA_type=T)genes=RNA_seq[,1]RNA_seq_exp<-RNA_seq[,3:ncol(RNA_seq)]RNA_seq_exp=data.matrix(RNA_seq_exp)rownames(RNA_seq_exp)<-genesRNA_seq_exp=avereps(RNA_seq_exp)RNA_seq_exp=as.data.frame(RNA_seq_exp)fwrite(file = paste0("TCGA_",namestype,"_ALL.txt"),RNA_seq_exp,sep = "\t",quote = F,row.names = T)lapply(c("protein_coding","lncRNA"),function(startype){RNAtype <- rownames(RNA_seq[RNA_seq[,"type"] %in% startype,])namesRNA<-ifelse(startype=="protein_coding","mRNA","lncRNA")RNA_seq_RNAtype=RNA_seq_exp[RNAtype,]fwrite(file = paste0("TCGA_",namestype,"_",namesRNA,".txt"),RNA_seq_RNAtype,sep = "\t",quote = F,row.names = T)})})
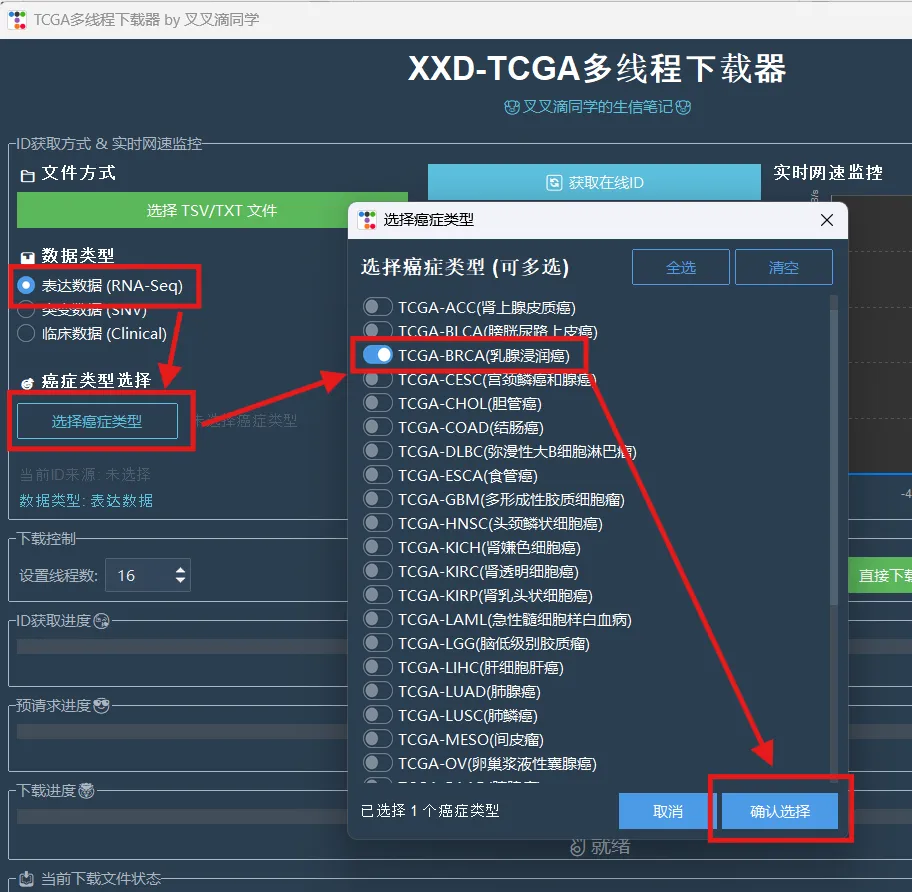
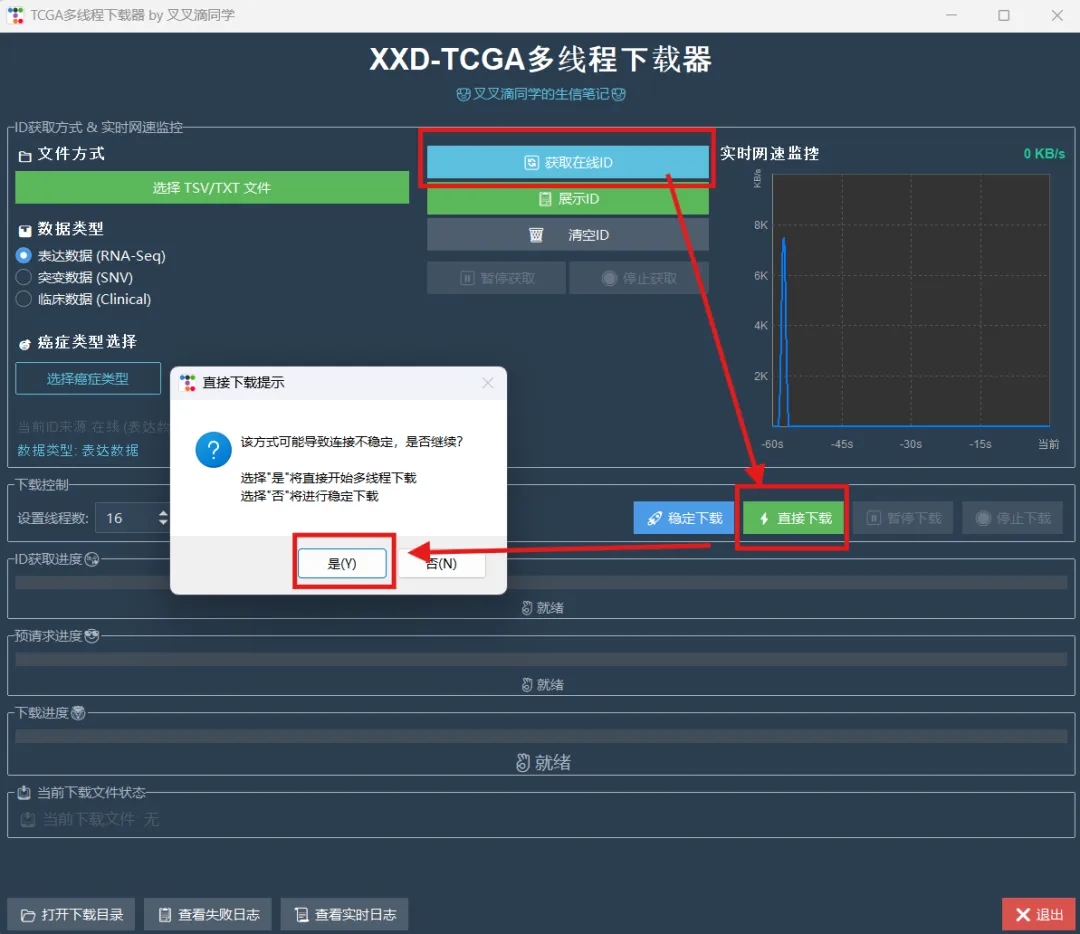
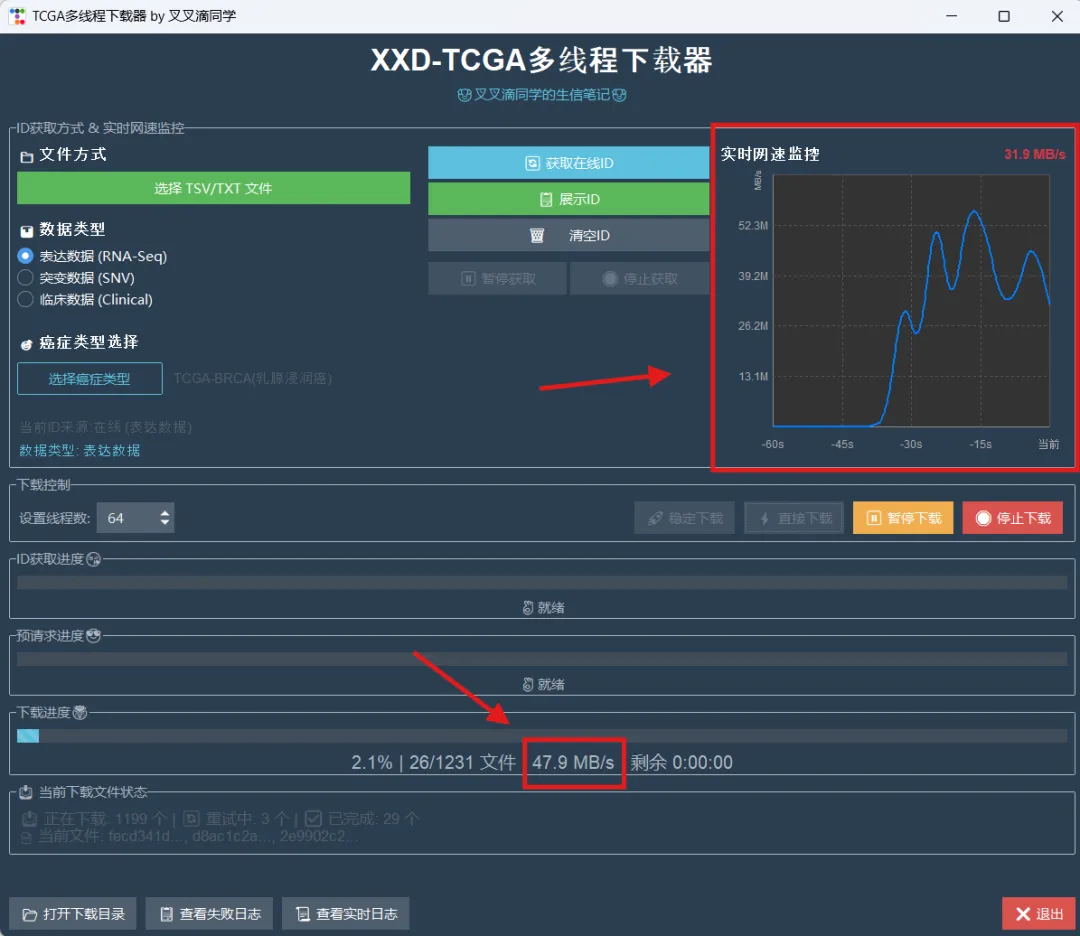
library(limma)library(future.apply)library(pbapply)library(parallel)library(doParallel)library(data.table)setDTthreads(threads =detectCores())source("xxdTCGAmerge.R",encoding = "utf-8")
tissue<-"TCGA-BRCA"metaMatrix.RNA=read.table("gdc_sample_sheet.2026-03-06.tsv",sep="\t",header=T)names(metaMatrix.RNA)=gsub("sample_id","sample",gsub("\\.","_",tolower(names(metaMatrix.RNA))))metaMatrix.RNA$sample_type=gsub(" ","",metaMatrix.RNA$tumor_descriptor)metaSample<-list.files(path = paste0(".\\GDC\\",tissue))samemeta<-intersect(metaMatrix.RNA$file_id,metaSample)metaMatrix.RNA<-metaMatrix.RNA[metaMatrix.RNA$file_id %in% samemeta,]
metaMatrix.RNA <- xxdTCGAmergeDuplicate(metaMatrix.RNA)metaMatrix.RNA <- xxdTCGAmergeSampleType(metaMatrix.RNA)
lapply(c("STAR","FPKM","TPM"),function(exptype){namestype<-ifelse(exptype=="STAR","Counts",ifelse(exptype=="FPKM","FPKM","TPM"))RNA_seq<-xxdTCGAmerge(metadata=metaMatrix.RNA,path=paste0(".\\GDC\\",tissue),data.type="RNAseq",mRNA_expr_type=exptype,symbol = T,RNA_type=T)genes=RNA_seq[,1]RNA_seq_exp<-RNA_seq[,3:ncol(RNA_seq)]RNA_seq_exp=data.matrix(RNA_seq_exp)rownames(RNA_seq_exp)<-genesRNA_seq_exp=avereps(RNA_seq_exp)RNA_seq_exp=as.data.frame(RNA_seq_exp)fwrite(file = paste0("TCGA_",namestype,"_ALL.txt"),RNA_seq_exp,sep = "\t",quote = F,row.names = T)lapply(c("protein_coding","lncRNA"),function(startype){RNAtype <- rownames(RNA_seq[RNA_seq[,"type"] %in% startype,])namesRNA<-ifelse(startype=="protein_coding","mRNA","lncRNA")RNA_seq_RNAtype=RNA_seq_exp[RNAtype,]fwrite(file = paste0("TCGA_",namestype,"_",namesRNA,".txt"),RNA_seq_RNAtype,sep = "\t",quote = F,row.names = T)})})

链接: https://pan.baidu.com/s/1zcbfxd3Tdo5l4PeQti8WQw?pwd=mtxs 提取码: mtxs

无法复制链接的同学可点击左下角的“阅读原文”访问网盘
