 夜雨聆风
夜雨聆风在生信分析中,很多时候如果拿不到基因表达矩阵或者作者处理后的数据不符合我们的要求,此时就需要去下载原始的测序数据(对于RNA-Seq来说, 通常是fastq格式)比对到参考基因组,从而生成下游分析用到的基因表达文件。
一般来说,对于GEO上的数据,其原始的测序数据基本上是存储在SRA(Sequence Read Archive)数据库,SRA数据库保存着由二代高通量测序产生的“短读序列”,通常小于1000 bp。
本文介绍使用sratoolkit工具下载SRA上的数据。
SRA数据的层级结构
在正式下载数据之前,还需要先了解 SRA 数据库中几个常见的编号体系,因为它们分别对应着不同层级的信息。
最上层通常是 BioProject,可以理解为一个完整研究项目的总入口,用来概括某项研究的整体设计与背景;一个 BioProject 往往会关联多个样本和多组测序数据。
再往下是 BioSample,它对应的是具体的生物学样本,也就是某一个实际采集到的组织、细胞或个体材料,每个独立样本都会有自己唯一的编号和属性信息。
对于测序本身来说,Experiment 表示的是针对某个样本构建的一套特定测序文库,反映的是实验设计层面的信息;而 Run 则对应真正产生的数据文件,可以把它理解为一次具体上机测序后的数据记录。
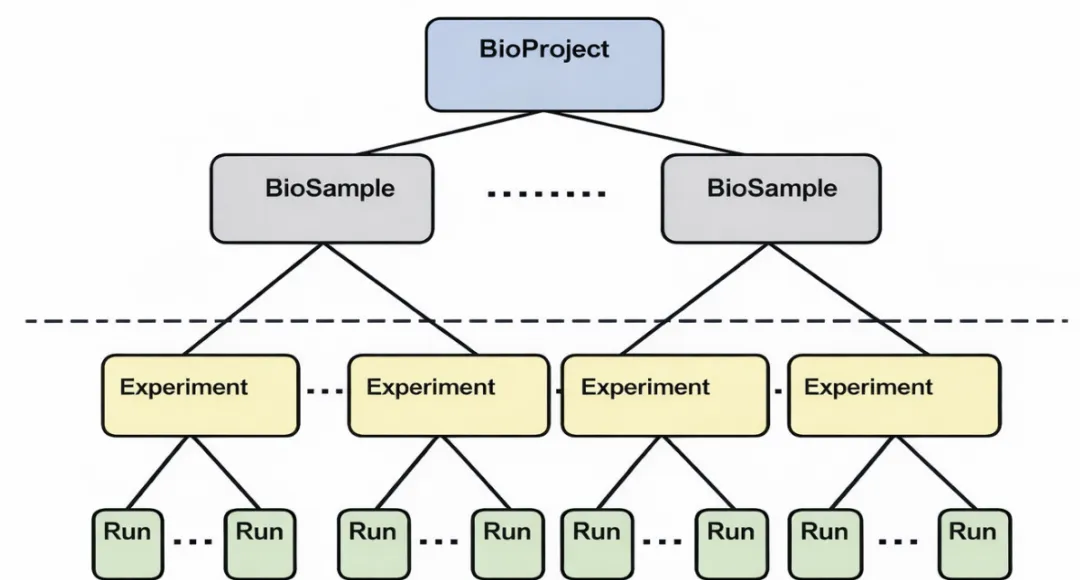
通常在实际下载原始数据时,我们最常接触、也最常使用的就是Run这一层级,因为它直接指向可下载的测序数据文件。
1. 获取SRR序号
在使用 SRA Toolkit 下载数据之前,首先要做的是在 SRA 数据库中找到目标数据对应的 Run accession,也就是我们常说的 SRR 编号。
这个编号是后续下载原始测序数据时最直接、最常用的入口。
具体来说,可以先在 NCBI 的 SRA 检索页面中,根据研究对象和测序类型设置关键词进行搜索。比如,如果我们想查找 BALB/c 小鼠淋巴结组织的 RNA-Seq 数据,就可以在检索框中输入相应的组合条件,将物种、品系、组织类型和测序策略一并限定,例如((("mus musculus"[Organism]) AND BALB/c*) AND "lymph*") AND "rna seq"[Strategy].。
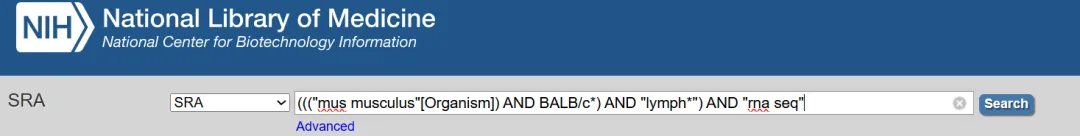
这样做的目的是尽量缩小结果范围,提高检索结果的针对性。
检索结果出来后,页面通常会显示一系列符合条件的实验记录。此时可以根据自己的研究目的,勾选感兴趣的记录;如果不手动勾选,系统一般默认导出当前检索结果中的全部记录。
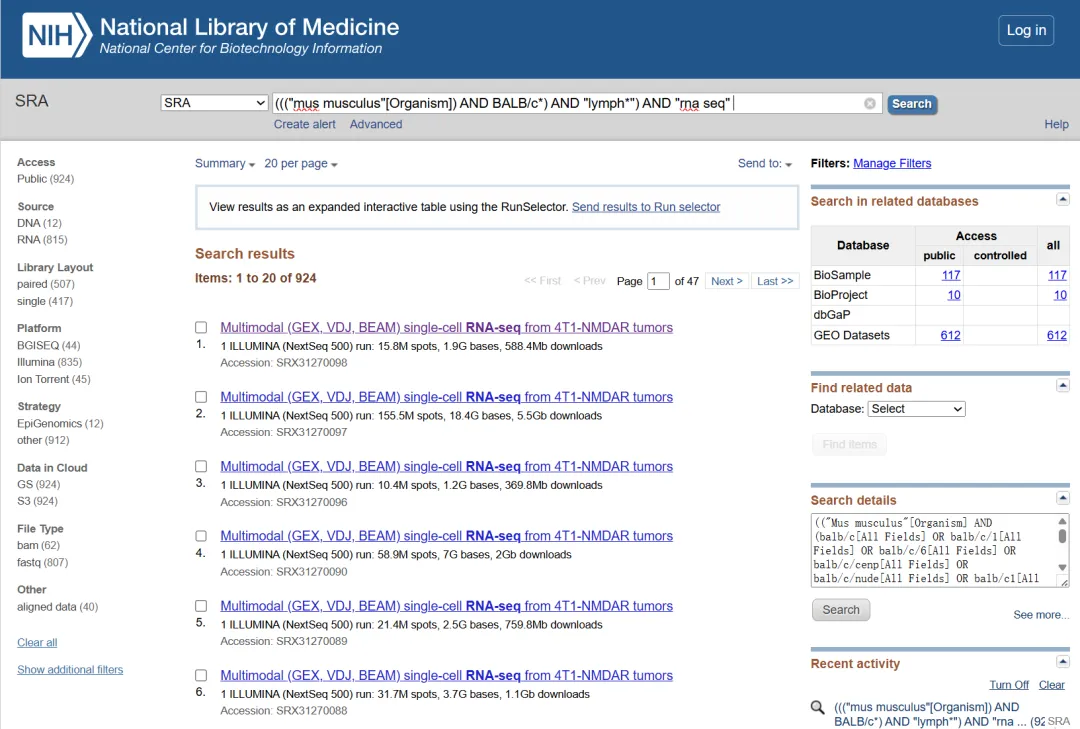
接下来,选中具体的条目,在页面上方选择 Send to,输出方式选择 File,文件类型选择 Accession List,即可将这些记录对应的 accession 列表保存下来。
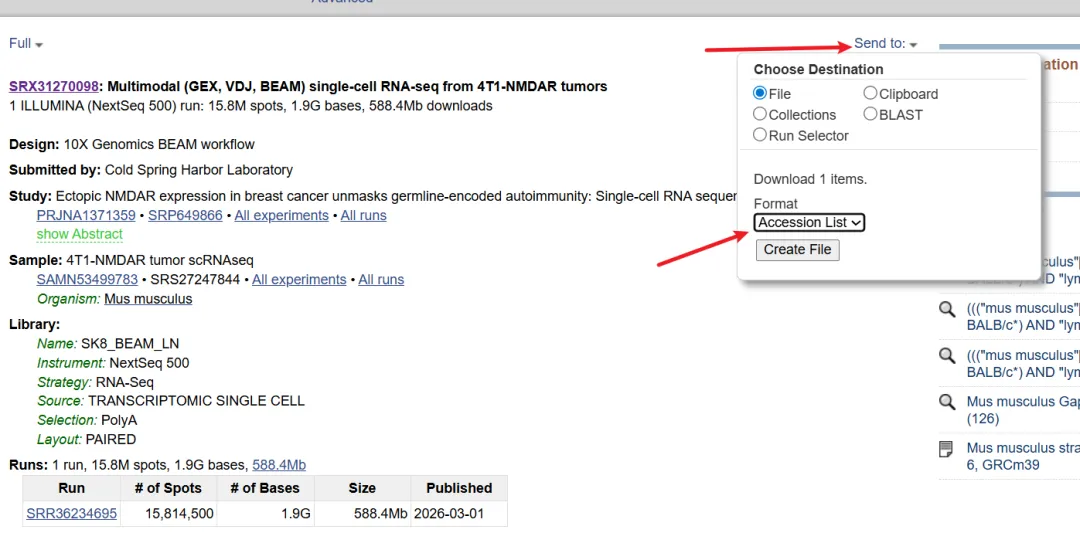
这里需要注意,真正用于下载 SRA 数据的并不是 BioProject 或 BioSample 编号,而是 Run accession。因此,导出的列表文件中通常是一行一个 SRR 编号,例如 SRR11192680、SRR11192681 这样的格式。
后续在使用 SRA Toolkit 批量下载数据时,往往就是直接读取这个 accession list 文件来完成下载。因此,这一步本质上就是先把需要的数据样本整理成一个可供程序调用的 SRR 清单。
2.下载SRA Toolkit
SRA Toolkit是 NCBI 官方提供的一套命令行工具,主要用于下载、解析和转换 SRA 数据,基本上是处理 SRA 原始测序文件时最常用的工具之一。
目前它提供了多个操作系统版本,包括 Ubuntu、Alma Linux、Mac OS X 以及 Windows,用户可以根据自己的系统环境选择对应的安装包进行下载。
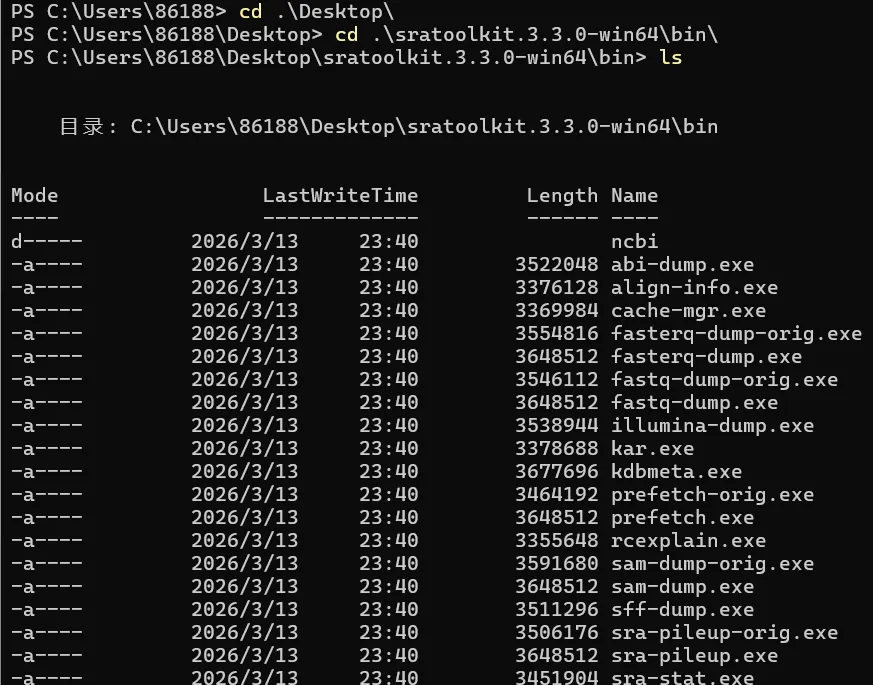
如果是在 Windows 系统下使用,安装过程相对直接。一般来说,只需要先下载对应的压缩包文件,然后将其解压到本地目录,例如桌面。
接着打开命令行窗口,进入解压后的文件夹,再切换到其中的 bin 目录,后续的大多数命令也都是在这个目录下执行,但要注意后面下载的测序数据最好不要放在这个目录。
具体安装步骤可见:https://github.com/ncbi/sra-tools/wiki/02.-Installing-SRA-Toolkit。
3. 测序数据下载
接下来就可以正式开始下载原始测序数据了。
在 SRA Toolkit 中,最常用的命令之一是 prefetch。它的作用是先把 SRA 数据库中的原始 Run 文件下载到本地,这些文件通常以压缩的 .sra 格式保存。
除了下载主体数据之外,prefetch 还会一并获取后续格式转换所需的相关信息,因此通常被视为下载 SRA 数据的标准步骤。对于下载过程中意外中断的任务,prefetch 也支持继续补全,这在批量下载时尤其有用。
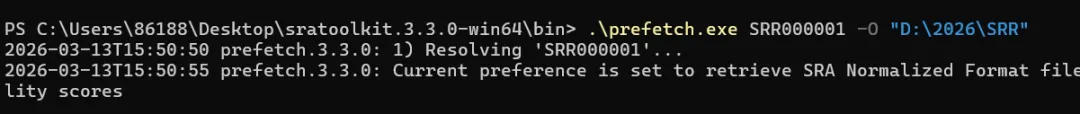
如果只需要下载单个样本,可以直接在命令后面跟上一个 SRR 编号,注意通过参数-O指定输出路径,否则会下载在当前目录:
prefetch SRR000001 -O <output-directory>
如果需要批量下载多个样本,则可以提前将所有 SRR 编号整理到一个文本文件中,再通过参数指定该文件,程序就会按照列表逐个下载:
prefetch --option-file SraAccList.txt -O <output-directory>
对于 RNA-Seq 这类项目来说,这种批量方式往往更高效,也更适合后续统一管理。
需要注意的是,prefetch 下载下来的仍然是 SRA 原始压缩格式,这种格式并不能直接用于常规的比对或定量分析。
因此,下载完成后,通常还需要借助 SRA Toolkit 中的另一个工具,例如 fasterq-dump,将 .sra 文件进一步转换为更常用的 FASTQ 格式(注意输入和输出文件的目录):
fasterq-dump --split-files SRR000001 .sra -O <output-directory>
如果是双端测序数据,还可以通过相关参数将两个方向的 reads 分别输出成独立文件,便于后续比对到参考基因组。
fasterq-dump --split-files SRR11180057.sra -O <output-directory>
在某些情况下,也可以跳过 prefetch 这一步,直接使用 fasterq-dump 输入 SRR 编号,一边下载一边完成格式转换。也就是说,数据获取和格式转换可以合并成一步操作。这样的方式在处理少量样本时比较方便,但如果数据量较大,或者网络环境不够稳定,通常还是建议先用 prefetch 完整下载,再单独进行格式转换,这样流程会更稳妥,也更便于排查问题。
