 夜雨聆风
夜雨聆风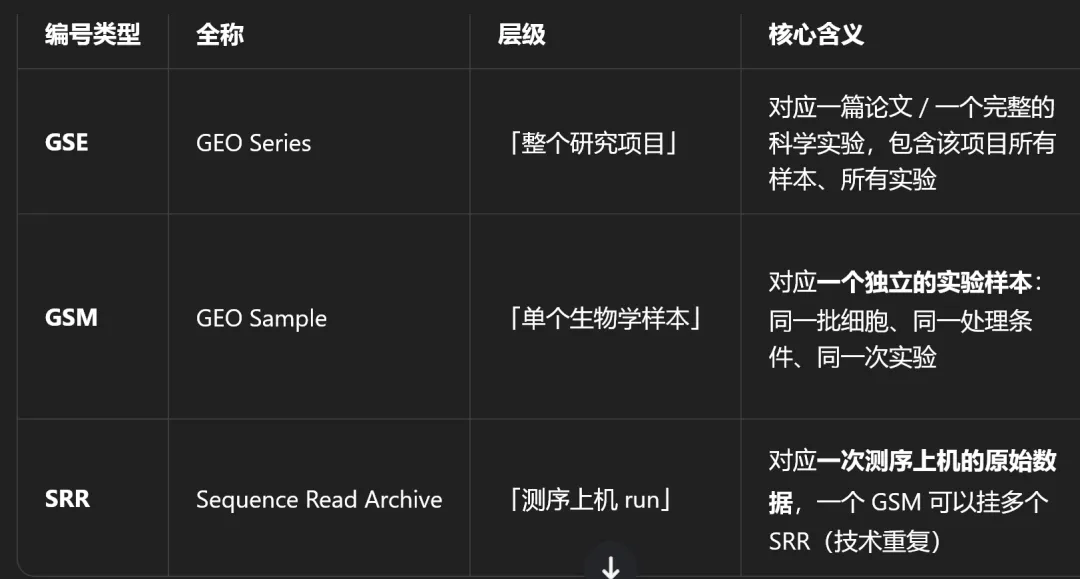
如何判断是生物学重复或者技术重复?
·查看 SRA 或 GEO 页面:
o同一个 GSMxxxxxx 下列出的多个 SRR,通常属于同一个样本的不同测序 run(技术重复)。
o如果它们是生物学重复,一般会有独立的 GSM 编号(例如 GSM1_rep1、GSM1_rep2)。
·查看文章方法部分:会说明每个 ChIP 样本是否有多个测序 lane。
数据分析建议:做 ChIP-Seq 分析时,通常会把同一个 GSM 下的两个 SRR 数据合并,作为该对照组的完整测序数据进行后续分析(如质控、比对、peak calling)。
# 合并两个SRR的fastq文件(假设已经下载好) cat SRR36732730.fastq SRR36732731.fastq > DMSO_FOXA1_ChIP_combined.fastq
一、数据库准备
C.实际操作流程(以两个NCBI SRA 为例):
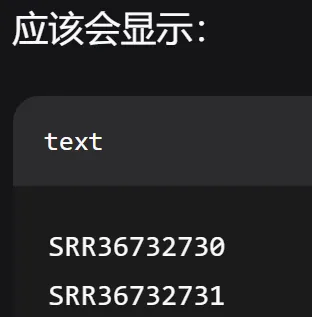
#打开终端,用 echo 命令创建一个 SRR_Acc_List.txt 文件
echo -e "SRR36732730\nSRR36732731" > SRR_Acc_List.txt
#用 cat 命令检查一下文件内容是否正确
cat SRR_Acc_List.txt
#使用 prefetch 命令配合 --option-file 参数,可以一次性读取列表中的所有 SRR 号并开始下载
prefetch --option-file SRR_Acc_List.txt
#批量转换为 FASTQ(单端专用命令), -e是线程数(建议不要超过逻辑线程)
for srr in $(cat SRR_Acc_List.txt); do
fasterq-dump$srr -e 16
done
#双端测序转换为FASTQ文件(--split-files:此参数会将双端测序数据自动分割为 _1.fastq 和 _2.fastq 两个文件)
for srr in $(cat SRR_Acc_List.txt); do
fasterq-dump$srr --split-files -e 16
done
二、质控
mkdir qcOutdir
fastqc SRR36732730.fastq SRR36732731.fastq -t 16 -o qcOutdir
cd qcOutdir
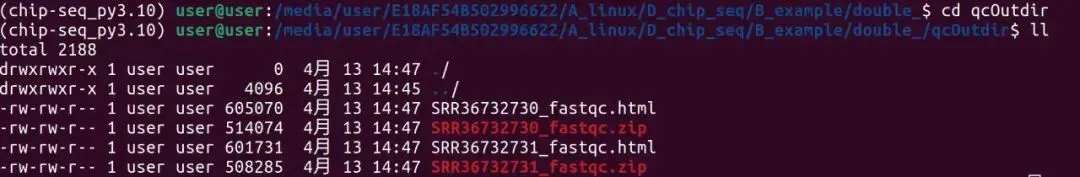
firefox SRR36732731_fastqc.html
#过滤
过滤主要有三步
#A. 去除adaptor序列
for sample in SRR36732730 SRR36732731; do
cutadapt-a AGATCGGAAGAGCACACGTCTGAACTCCAGTC \
${sample}.fastq\
-m30 -j 18 \
--output=${sample}.cutAd1.fq
done
#B. 去除N序列 (removeN是个脚本,读取一个FASTQ文件,计算每条read中 N或n(未确定碱基)占该read总长度的比例。
如果N比例 ≤ 5%(默认),这条read写入 *. unknowNul1.fq(好reads)
如果 N比例 > 5%,则写入 *.N.fq(N含量高的reads))
输出的文件SRR36732730.unknowNul1.fq是合格的reads(N比例<5%)
输出的文件SRR36732730. cutAd1.N.fq 是不合格reads (N比例>5%)
for sample in SRR36732730 SRR36732731; do
./removeN${sample}.cutAd1.fq ${sample}.unknowNul1.fq
done
#C去除低质量reads
# basename:这是一个 Linux 命令,功能是从一个完整的文件路径中提取出文件名,并可以去掉指定的后缀。
# f:循环变量,依次代表列表中的每个原始文件名。第一次循环:f = SRR36732730.unknowNul1.fq
for f in SRR36732730.unknowNul1.fq SRR36732731.unknowNul1.fq; do
base=$(basename$f .unknowNul1.fq)
fastp-i $f -o ${base}.clean1.fq \
--qualified_quality_phred20 --unqualified_percent_limit 30 \
--thread16
done
#查看质控结果
for f in SRR36732730.clean1.fq SRR36732731.clean1.fq; do
fastqc$f -t 16 -o qcOutdir
done
三、对比
# 即mapping需要的软件:software: bowtie2 (bowtie2) + samtools (samtools)
conda install bowtie2 samtools
#这个命令试图用 Bowtie2 将过滤后的测序 reads 比对到 人类参考基因组 hg19 上,生成一个 SAM 格式的比对文件。-p 16:使用 16个 CPU 线程 。-U(单端)或 -1/-2(双端)明确指定。
for f in SRR36732730.clean1.fq SRR36732731.clean1.fq; do
base=$(basename$f .clean1.fq)# 提取 SRR36732730 或 SRR36732731
bowtie2 -p 16 -x /media/user/E18AF54B502996622/A_linux/D_chip_seq/A_hg19/hg19 -U $f -S ${base}.sam
done
#只留下唯一比对,去除多余比对
#先查看sam文件
less SRR36732730.sam

#去多余比对
·-b:输出 BAM 格式(二进制压缩格式),而不是默认的 SAM 文本格式。 不加 -b 时,samtools view 输出 SAM 文本;加上 -b 则输出 BAM。
·-S:指明输入是 SAM 格式(而不是 BAM)。@后面是线程数
for sample in SRR36732730 SRR36732731; do
samtoolsview -h -@ 16 ${sample}.sam | grep -v 'XS:i:' | samtools view -bS -@ 16 - > ${sample}.unique.bam
done
#打开这个bam文件,查看头文件
samtools view -h SRR36732730.unique.bam | grep -c 'XS:i:'
#然后查看一下还有没有XS,这个代码出结果,最后面应该是0
四、call peak
#需要使用的软件macs3
conda install macs3
#call peak (peak finding) : -t 实验组;-c对照组;-f输入文件类型;-g 染色体选择的物种或者染色体长度(非模式物种);-n 输出一大堆结果的头文件;-p p值的过滤条件;-B 输出bed文件
#用下面这个
macs3 callpeak -t Treatment_combined.unique.bam -c DMSO_combined.unique.bam -f BAM -g hs -n sample -p 1e-5 -B
#查看bed文件,按q键退出
less -SN sample_summits.bed
#主要是要查看xls文件
le sample_peaks.xls
#如果peaks还是非常多,那么后面也可以根据FDR值进行过滤
#查看summits.bed文件,会告诉我们每个peak的最高峰
# ,是实验组在某个区域比较低,对照组在这个区域反而是比较高的
# sample_peaks.narrowPeak 是 MACS2 / MACS3 输出的核心结果文件
#下面是peak的画图
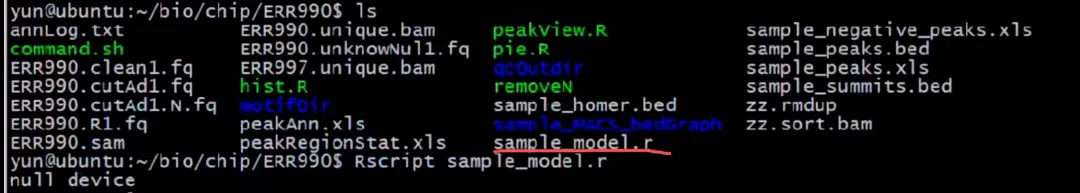
#根据MACS3自己生成的R文件先画图,完事后会生成,sample_model.pdf这个文件
Rscript sample_model.r
#然后查看这个生成的pdf图
evince sample_model.pdf #双峰交汇处是正确的结合位点
#查看行号,为了是hist.R文件查看的时候去除多余的行,然后可以改峰的长度,更贴合xls表的数据
less -N sample_peaks.xls
#主要是需要根据sample_peaks.xls修改这个R脚本的行号。生成的是peakHist.pdf文件,看peak的长度
Rscript hist.R
evince peakHist.pdf
#进行peakview,peak的可视化
#得到的两个bdg文件,分别是treat和control。
#要修改peakView.R的具体参数
Rscript peakView.R
#Homer软件进行peak注释
conda install homer
#查看需要下载的注释基因组类型,如果没有,也可以自己设定
perl /home/user/miniconda3/envs/chip-seq_py3.10/share/homer/configureHomer.pl -list
#同时还要安装基因组注释软件,bowtie2用哪个,现在的注释软件就用哪个#同时还有四个依赖软件
conda install ghostscript weblogo ucsc-blat samtools
#bed文件的输出的sample_peaks.narrowPeak转为homer的输入
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' sample_peaks.narrowPeak > sample_homer.bed
less -N sample_homer.bed
#安装基因组
perl /home/user/miniconda3/envs/chip-seq_py3.10/share/homer/.//configureHomer.pl -install hg19
#找motif。按照8,10,12的长度去寻找。motifDir是新建的文件夹存放文件的。
findMotifsGenome.pl sample_homer.bed hg19 motifDir -len 8,10,12
#已知基序富集:knownResults.html 相关文件,帮你回答「我的 ChIP 峰里,哪些已知转录因子的结合基序显著富集?」
#从头基序预测:homerResults.html 相关文件,帮你回答「我的 ChIP 峰里,有没有新的、未被报道的转录因子结合基序?」
cd motifDir
cd ..
#对peak进行注释。1是注释结果,2是错误结果。目前没有好的方法多文件并行
annotatePeaks.pl sample_homer.bed hg19 1>peakAnn.xls 2>annLog.txt
#也可以对peak进行kegg和go注释(在GODir文件中)
annotatePeaks.pl sample_homer.bed hg19 -go GODir 1>peakAnn.xls 2>annLog.txt
less -N peakAnn.xls
#统计peakAnn.xls中,第8列的类型
awk -F "\t" 'NR>1{ split($8, arr, "("); sub(" ", "",arr[1]); print arr[1]}' peakAnn.xls | sort | uniq -c | awk '{print $2"\t"$1}' > peakRegionStat.xls
less -N peakRegionStat.xls
Rscript pie.R
evince peakRegionStat.pdf#查看peak存在哪些区域
