 夜雨聆风
夜雨聆风本期覆盖时间:2026.04.29-2026.05.12

封面图:AI 生成|组学与单细胞 AI 主题配图
导读
本期周二栏目继续聚焦“组学与单细胞 AI”。这两周的文章有一个很明显的变化:AI 不再只是给组学表格做分类,而是在尝试改写数据进入科研问题的方式。
最值得注意的是,空间组学和病理图像开始更紧地接在一起。Path2Space 用病理切片预测空间基因表达,把昂贵的空间转录组实验转成可以在大队列里扩展的推断流程。这对做组织结构、炎症微环境、肿瘤微环境或口腔组织空间异质性的人,启发很直接:图像不只是配图,它可能成为组学空间预测的入口。
另一条主线是单细胞基础模型正在变得更“懂生物”。RegFormer 把基因调控网络先验放入模型结构,CAPTAIN 则直接用 RNA 和蛋白共测数据做多模态预训练。它们都在回答同一个问题:细胞状态不能只靠转录本描述,模型需要看见调控层级、蛋白层读数和跨模态依赖。
本期按 7 篇文章展开:前四篇作为主文,分别对应空间表达预测、单细胞调控基础模型、RNA/蛋白多模态模型和增强子-基因调控连线;后三篇作为延伸阅读,补上空间多组学整合、单细胞/空间蛋白组融合和 scATAC 细胞类型注释。
本期速览
先用一张图看本期 7 篇文章的结构:从图像到空间表达,从转录组到蛋白层,从细胞注释到调控网络,AI 正在把“多模态数据”变成可推断、可迁移、可复用的科研流程。
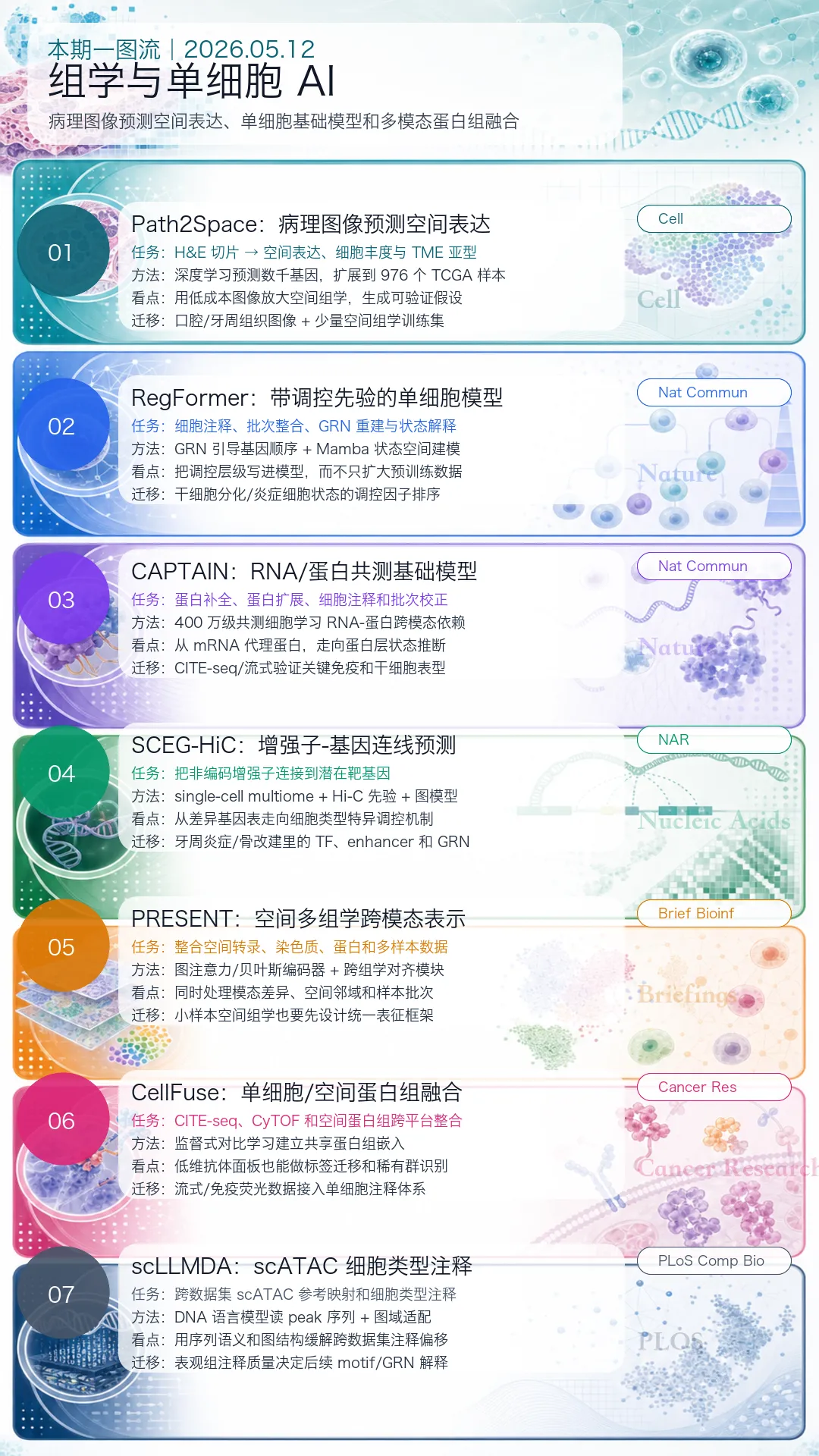
本期正式解读如下。
01|Path2Space:病理图像不只是配图,也可以预测空间基因表达
研究背景:空间转录组能把基因表达放回组织结构里,但成本和通量限制了它在大样本队列中的应用。病理切片几乎是很多队列都已有的数据,如果能从 H&E 图像预测空间表达,就有机会把空间组学问题带到更大的样本规模。
方法亮点:作者提出 Path2Space,用深度学习模型从乳腺癌病理图像直接预测空间基因表达。模型在空间转录组数据上训练,并用于推断大量 TCGA 乳腺癌样本的空间肿瘤微环境特征。文章还把预测表达进一步转成细胞类型丰度、空间亚型和治疗反应相关指标。
主要结论:Path2Space 可以较稳定地预测数千个基因的空间表达,并从 976 个乳腺癌样本中推断出具有不同预后和治疗反应特征的空间微环境亚型。它的核心价值不是替代空间转录组实验,而是在已有病理图像中生成可筛选的空间组学假设。
启发与迁移:这篇非常贴近“影像 + 组学 + 机制”的迁移方向。做口腔黏膜、牙周炎症、骨缺损修复或肿瘤微环境时,如果已有组织图像和少量空间组学训练数据,类似思路可以帮助我们先在大样本图像中定位空间表达趋势,再回到实验里做验证。
▍文献信息英文标题:AI-predicted spatial transcriptomics unlocks breast cancer biomarkers from pathology期刊/平台:Cell第一作者:Eldad D. Shulman通讯作者:Eldad D. Shulman 等单位:Cancer Data Science Laboratory, National Cancer Institute;University of Maryland;Sungkyunkwan University 等DOI:10.1016/j.cell.2026.04.023 公开资源:代码和教程:https://zenodo.org/records/14729337;论文 DOI:https://doi.org/10.1016/j.cell.2026.04.023 来源链接:https://pubmed.ncbi.nlm.nih.gov/42105763/
02|RegFormer:单细胞基础模型开始把调控网络写进模型结构
研究背景:单细胞大模型的一个常见问题是,模型可能学到表达模式,却不一定理解基因之间的调控层级。对机制研究来说,只会聚类和注释还不够,我们更关心哪些基因调控关系在细胞状态变化中起作用。
方法亮点:RegFormer 将基因调控网络先验和 Mamba 状态空间模型结合起来。它不是把基因当作无序 token,而是按调控层级组织基因顺序,同时为每个基因编码表达值和调控身份。模型在 2500 万个人类单细胞上预训练,覆盖多组织和多类生物学场景。
主要结论:研究显示,RegFormer 在细胞聚类、批次整合、细胞类型注释和调控网络重建任务中都有较强表现。更重要的是,它尝试把“表达矩阵学习”推进到“调控结构学习”,这让单细胞基础模型更接近机制挖掘工具。
启发与迁移:如果后续要做牙周炎症细胞状态、干细胞分化或骨改建相关单细胞图谱,RegFormer 的启发是:不要只问某个 cluster 的 marker 是什么,还要问这些 marker 背后的调控层级是否能被模型重建。它也适合作为后续“虚拟扰动/GRN 工作流”的候选。
▍文献信息英文标题:RegFormer: a single-cell foundation model powered by gene regulatory hierarchies期刊/平台:Nature Communications第一作者:Luni Hu通讯作者:Hansheng Zhao;Shuangsang Fang;Yuxiang Li;Yong Zhang单位:BGI Research;Chinese Academy of Agricultural Sciences;Chinese Academy of Sciences 等DOI:10.1038/s41467-026-72198-x 公开资源:论文页:https://www.nature.com/articles/s41467-026-72198-x;独立代码入口待核验来源链接:https://pubmed.ncbi.nlm.nih.gov/42086551/
03|CAPTAIN:单细胞模型不能只看 RNA,也要补上蛋白层
研究背景:转录组是最常见的单细胞数据,但蛋白才更接近细胞功能输出。很多细胞状态、免疫表型和治疗相关特征不能只靠 mRNA 可靠替代,因此 RNA/蛋白共测数据正在成为单细胞多模态建模的重要训练材料。
方法亮点:CAPTAIN 是一个在共测单细胞 RNA 和表面蛋白数据上预训练的多模态基础模型。它使用超过 400 万个细胞和 382 个表面蛋白,学习 RNA 与蛋白之间的跨模态依赖,可用于蛋白补全、蛋白扩展、细胞类型注释和批次校正。
主要结论:研究显示,CAPTAIN 能在 fine-tuning 和 zero-shot 场景中保持较好的泛化能力。它的价值不只是提高某个 benchmark 分数,而是把单细胞模型从“只会读转录组”推向“能同时理解 RNA 和蛋白表型”的方向。
启发与迁移:对免疫微环境、牙周炎症和干细胞状态研究来说,这篇提醒我们:没有蛋白层数据时,AI 可以做补全,但补全结果必须被当作推断,而不是实测。更理想的路线是,在关键问题上设计少量 CITE-seq 或流式验证,把模型预测和真实蛋白读数闭环。
▍文献信息英文标题:CAPTAIN: a multimodal foundation model pretrained on co-assayed single-cell RNA and protein期刊/平台:Nature Communications第一作者:Boya Ji通讯作者:Shaoliang Peng;Fulong Yu单位:Hunan University;Guangzhou Medical University;Central South University 等DOI:10.1038/s41467-026-72882-y 公开资源:论文页:https://www.nature.com/articles/s41467-026-72882-y;代码:https://github.com/iamjiboya/CAPTAIN来源链接:https://pubmed.ncbi.nlm.nih.gov/42098152/
04|SCEG-HiC:从单细胞多组学推断增强子和靶基因连线
研究背景:很多疾病相关变异位于非编码区域,真正困难的问题是:这些增强子到底调控哪些基因。单细胞 ATAC/RNA 数据能提供细胞类型特异的开放染色质和表达信息,但缺少空间构象信息时,增强子-基因连线仍然容易不稳。
方法亮点:SCEG-HiC 使用 weighted graphical lasso,将单细胞 multiome 数据和 bulk Hi-C 先验结合起来,预测增强子与靶基因之间的调控链接。它同时支持 paired scATAC/RNA-seq 和 scATAC-only 输入,因此更接近现实数据场景。
主要结论:文章在 10 个人和小鼠单细胞多组学数据集上评估,显示 SCEG-HiC 在增强子-基因链接预测上优于多种已有方法。作者还将其应用于 COVID-19 数据,展示了重建疾病严重度相关调控网络和连接非编码变异到潜在靶基因的能力。
启发与迁移:这篇适合放进“机制挖掘”工具箱。很多口腔基础和炎症研究最后都会回到一个问题:差异基因背后由谁调控,非编码区域是否解释了细胞状态差异。SCEG-HiC 提供的是从单细胞多组学走向调控机制的一条路线。
▍文献信息英文标题:Predicting enhancer-gene links from single-cell multi-omics data by integrating prior Hi-C information期刊/平台:Nucleic Acids Research第一作者:Xuan Liang通讯作者:Zhen Wang单位:Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences;Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences 等DOI:10.1093/nar/gkag437 公开资源:论文页:https://academic.oup.com/nar/article/54/9/gkag437/8672760;独立代码入口待核验来源链接:https://pubmed.ncbi.nlm.nih.gov/42100854/
延伸阅读|三篇也值得保存
05|PRESENT:空间多组学整合要同时处理模态差异和样本差异
研究背景:空间组学已经不只是空间转录组,越来越多技术同时测量基因、染色质、蛋白或其他分子层。真正困难的地方在于,不同模态的噪声、尺度和空间分辨率不一致,不同样本之间还存在批次和组织差异。
方法亮点:PRESENT 是一个面向空间多组学的对比学习整合框架。它使用图注意力网络、贝叶斯神经网络和分布感知解码器为不同组学模态建立编码器,再通过跨组学对齐模块整合空间信息和多模态信息。
主要结论:研究显示,PRESENT 可以用于空间 domain 识别、跨模态表示和多样本整合。它的意义在于把空间多组学从“各看各的数据层”推向“在同一空间表征里比较组织微环境”。
启发与迁移:如果未来做口腔组织或骨组织的空间组学,样本量通常不会大,模态也可能不完整。PRESENT 的思路提醒我们,空间邻域、组学模态和样本批次最好一开始就放入统一设计,而不是最后再靠可视化硬拼。
▍文献信息英文标题:Cross-modality representation and multi-sample integration of spatially resolved omics data期刊/平台:Briefings in Bioinformatics第一作者:Zhen Li通讯作者:Rui Jiang;Lei Zhai单位:Tsinghua University;Harbin Institute of Technology;Nankai University 等DOI:10.1093/bib/bbag214 公开资源:代码:https://github.com/lizhen18THU/PRESENT;论文 DOI:https://doi.org/10.1093/bib/bbag214 来源链接:https://pubmed.ncbi.nlm.nih.gov/42114120/
06|CellFuse:单细胞和空间蛋白组也需要自己的整合模型
研究背景:很多单细胞整合工具默认输入是转录组,或者假设不同数据集之间有大量共同特征。但蛋白组,尤其是抗体面板、CyTOF、CITE-seq、CODEX、IMC 和 MIBI-TOF,经常只有较少重叠 marker,直接套转录组整合方法容易失真。
方法亮点:CellFuse 是一个面向抗体蛋白组数据的深度学习整合框架。它使用监督式对比学习建立共享嵌入空间,用于跨平台细胞类型预测、标签迁移和数据整合,覆盖悬液单细胞蛋白组和空间蛋白组场景。
主要结论:研究显示,CellFuse 能在外周血、骨髓、淋巴瘤和实体瘤数据中实现较稳的跨模态细胞类型预测,并能帮助恢复临床相关的稀有细胞群。它的重点不是“多加一个模型”,而是承认蛋白组数据有自己的稀疏性和面板差异。
启发与迁移:这对口腔炎症、免疫微环境和组织修复研究很有用。很多实验室更容易拿到流式、CITE-seq 或免疫荧光/空间蛋白数据,而不是完整单细胞多组学。CellFuse 代表了一种把低维蛋白面板做成可迁移注释体系的路线。
▍文献信息英文标题:CellFuse enables Multi-modal Integration of Single-cell and Spatial Proteomics Data for Systems-level Analysis in Cancer期刊/平台:Cancer Research第一作者:Abhishek Koladiya通讯作者:Abhishek Koladiya;Kara L. Davis单位:Stanford University School of Medicine;Stanford Cancer Institute 等DOI:10.1158/0008-5472.CAN-25-3699 公开资源:代码:https://github.com/karadavis-lab/CellFuse;复现资料:https://zenodo.org/records/18088974来源链接:https://pubmed.ncbi.nlm.nih.gov/42084223/
07|scLLMDA:scATAC 注释开始借用 DNA 语言模型
研究背景:scATAC-seq 能告诉我们染色质开放状态,但细胞类型注释比 scRNA-seq 更难。常见做法是用 scRNA-seq 做跨模态标签转移,但 ATAC 和 RNA 之间存在模态差异,容易引入信号偏移。
方法亮点:scLLMDA 使用预训练 DNA 语言模型提取 peak 序列语义,再结合 accessibility 信息表示单个细胞。随后,方法构建源域和目标域的细胞图,用图神经网络做 domain adaptation,在保留局部结构的同时完成 scATAC 细胞类型注释。
主要结论:文章显示,序列语言模型和图结构可以共同提升 scATAC 注释表现,尤其适用于跨数据集、跨条件的参考映射场景。它把“细胞类型注释”从简单 label transfer 推向了“序列特征 + 图结构 + 域适配”的组合。
启发与迁移:如果以后做表观组、染色质开放或单细胞多组学,scATAC 注释质量会直接影响后续 TF motif、增强子和调控网络解释。scLLMDA 的价值在于提醒我们:注释不是前处理小步骤,而是机制解释的地基。
▍文献信息英文标题:Cell type annotation for scATAC-seq via DNA large language model and graph domain adaptation期刊/平台:PLoS Computational Biology第一作者:Yan Liu通讯作者:Ji-Peng Qiang;Guo Wei单位:Yangzhou University;Nanjing Forestry University;Nanjing University of Science and Technology;Bengbu University 等DOI:10.1371/journal.pcbi.1014226 公开资源:论文页:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1014226;代码入口待核验来源链接:https://pubmed.ncbi.nlm.nih.gov/42060707/
本期小结
本期可以形成三个判断。
第一,空间组学 AI 正在和图像更深地融合。Path2Space 这类工作说明,组织图像可能成为空间表达预测、细胞组成推断和空间 biomarker 探索的入口。
第二,单细胞基础模型正在从“更大数据量”转向“更合理的生物先验”。RegFormer 引入调控网络,CAPTAIN 引入蛋白层共测,都是在补足单纯表达矩阵的盲点。
第三,真正值得保存的工具通常出现在中间流程:增强子-基因连线、空间多组学对齐、蛋白组跨平台注释、scATAC 注释。这些工具不一定最适合做标题,但最适合沉淀成自己的科研方法库。
