 夜雨聆风
夜雨聆风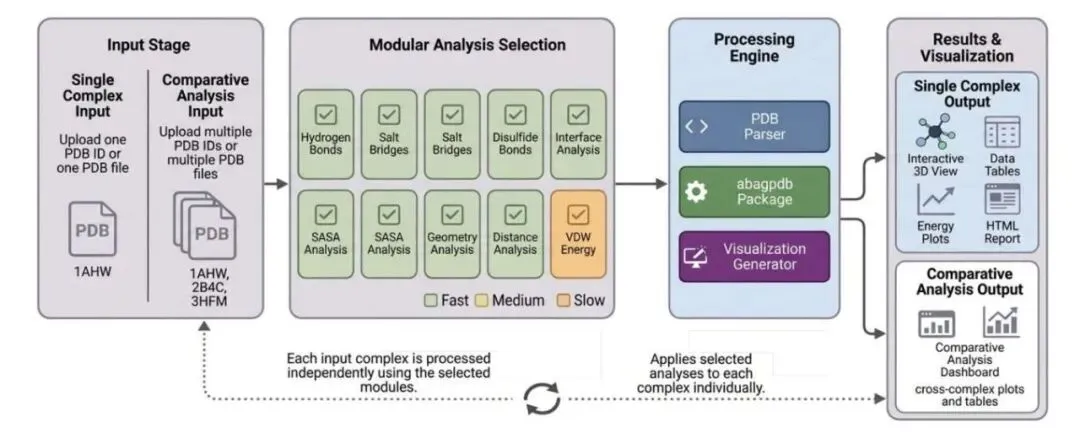
<<< 左右滑动见更多 >>>
榴莲忘返AIDD
供稿 | 柠檬青年
审稿 | 吉星
目录
SPICE 模块化整合了多项结构分析功能,让研发人员能在几秒内完成野生型与突变体等不同蛋白复合物的界面特征和能量差异对比。 BOLEK 将分子指纹压缩成单一 Token 嵌入大语言模型,并强制 AI 基于真实的化学描述符(如 LogP、TPSA)进行推理,彻底打破了分子预测的黑盒难题。 这是一套在超级计算机上运行的自主推理系统,它模拟科研团队的工作方式,去设计能靶向无序蛋白(IDP)的生物药。 蛋白质大语言模型提取的残基上下文特征在双序列比对中,实际效果超越了先用 AlphaFold3 预测再进行结构比对的策略。 DoFormer 将因果推断直接写入 Transformer 注意力机制,彻底抛弃了脱离生物学实际的有向无环图假设,让单细胞层面的未知基因扰动预测变得更加精准。

1. 抗体与蛋白研发利器:SPICE 秒级对比结构
做过蛋白 - 蛋白相互作用(PPI)优化或抗体工程的同行都知道,分析突变体和野生型复合物的结构差异是个纯纯的体力活。通常你需要分别跑软件计算氢键、提取溶剂可及表面积(SASA)、检查有没有空间位阻,最后把几堆乱七八糟的数据塞进 Excel 里自己对比。
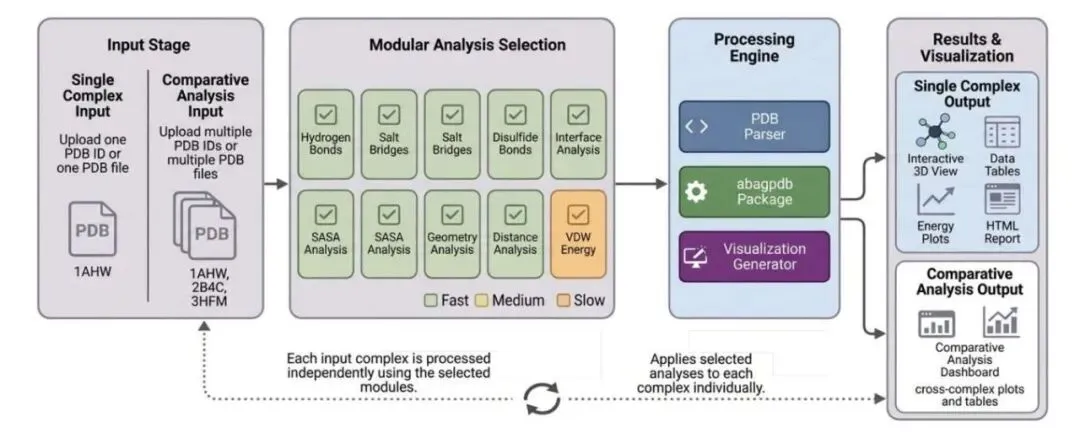
研究者开发的 SPICE(Structural Protein Interaction Complex Evaluator)网页平台解决了这个痛点。这工具直接读取 PDB 结构,把繁琐的对比工作自动化了。
勾选模块,几秒出结果
SPICE 采用了模块化设计。你需要算什么,打个勾就行。这种做法把原本需要切来切去的分析流程缩短到了几秒钟。跑出来的数据结构清晰,后续重跑或者复现也毫无压力。
为多复合物比对而生
这工具最实用的地方在于它把「对比」作为了一等公民。当你把野生型和突变体、或者不同的结合配体扔进去,它会直接汇总出差异表格和图表。
在残基层面,它把距离在 5.0 Å 内的氨基酸划分为界面残基。工具会统计每个位置的接触频率和类型,帮你快速锁定结合热点,看清特定残基在突变后发生了什么变化。它设定了明确的几何标准来识别氢键、盐桥和二硫键,保证跨复合物对比时的数据口径完全一致。
评估能量与几何质量
评估突变好坏,光看接触面积远远不够。SPICE 整合了 FreeSASA(使用 1.4 Å 探针的 Lee-Richards 算法)来计算溶剂可及表面积和埋藏表面积(BSA)。它还引入了 Lennard-Jones 6-12 势函数来估算残基间的范德华相互作用。这里给出的相对能量数值,能直观地告诉你某个突变带来了良好的局部堆叠,还是引发了糟糕的空间位阻。
几何评估方面,拉氏图(Ramachandran plots)是标配,SPICE 专门把界面残基在图上高亮了出来。它还附带了侧链构象分析、主骨架弯曲角度评估,以及带有可调阈值的残基间距离热图,方便排查结构上的别扭之处。
看清突变背后的驱动力
在对比多个复合物时,SPICE 支持并排同步操作的 3D 视图,你可以用 Biopython 模块先做个对齐。它生成的突变影响摘要极具参考价值,比如它会输出 ΔVDW 对 ΔSASA 的散点图。这类图表能帮你拆解结合力改变的原因:到底是由能量驱动的,还是由于几何构象改变带来的。
研究者用两个真实案例跑了测试。一个是经典的抗体 D1.3-溶菌酶(1A2Y),展示了单复合物的界面提取和范德华力映射。另一个更贴近实战:PD1 结合帕博利珠单抗(5GGS),对比野生型与一个单点突变体。借此可以清晰地定位突变如何引起局部埋藏面积、接触类型和能量的转移,配合 3D 视图一目了然。
📜Title: Comparative Structural Analysis of Protein Complexes With SPICE
🌐Paper: https://doi.org/10.1093/nar/gkag415
💻Code: https://github.com/fbabd/PyPdbComplex

2. 告别黑盒预测:BOLEK 让 AI 基于真实特征读懂分子
做过计算辅助药物发现(CADD)的人都有个共同的痛点。你把一个分子扔进机器学习模型,它吐出一个干巴巴的分数。你问它为什么是这个分数,现在流行的大语言模型(LLM)就开始胡说八道。它会编造一堆听起来专业的「机制解释」,但你根本没法对照真实的分子结构去验证。
研究者开发了 BOLEK 模型来解决这个问题。它的核心逻辑很简单:让模型的自然语言推理建立在可验证的分子特征上。
来看看底层的架构设计。研究者选用了 Qwen3-4B-Instruct 作为基础模型。他们做了一个关键改动,把一个 2048 位、半径为 2 的摩根指纹(Morgan fingerprint)通过一个小型投影器映射到 LLM 的嵌入空间里,变成一个单一的 token,随后进行端到端的训练。这就好比不仅给 AI 看了分子的名字,还直接递给它一张高度浓缩的结构特征扫描图。
训练对齐方式是这篇文章的亮点。很多多模态模型喜欢用大段的笼统描述性文字来训练。BOLEK 走的是「第一性原理」路线。研究者用了超过 85 万个分子,强迫模型回答具体的硬核问题。比如:这个具体的结构片段存在吗?具体的数值描述符是多少?
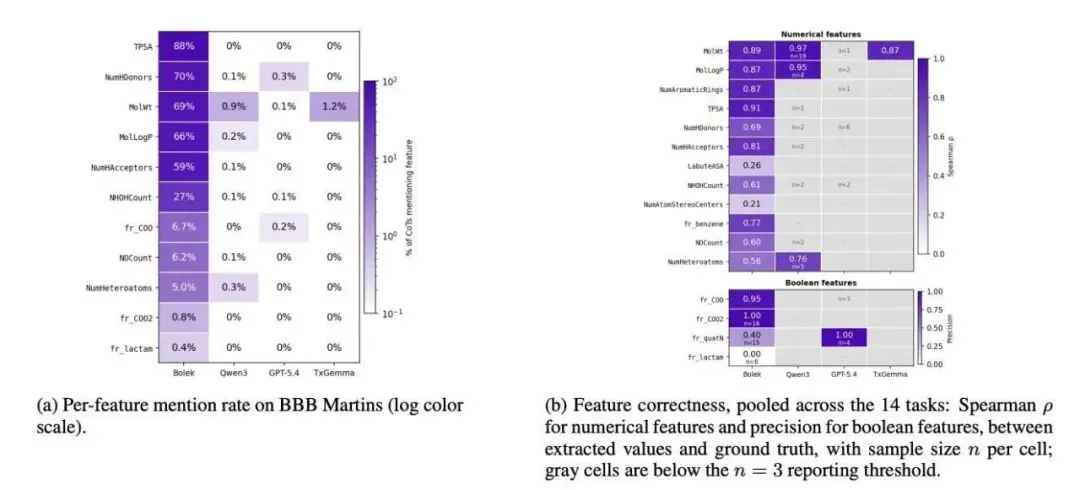
为了让模型真正学会推理,研究者在数据构建上花了不少心思。他们为 15 个 TDC(Therapeutics Data Commons)二分类任务生成了带有思维链(CoT)的训练数据,涵盖 hERG、BBB、AMES 等常见成药性终点。这个思维链是有现实锚点的。提示词里包含文献提取的机制说明、SMILES 字符串、局部结构注释,外加随机森林算法挑出来的排名前 20 的 RDKit 描述符数值。模型在做出「有毒」或「无毒」的判断前,必须先看这些真实的化学硬指标。
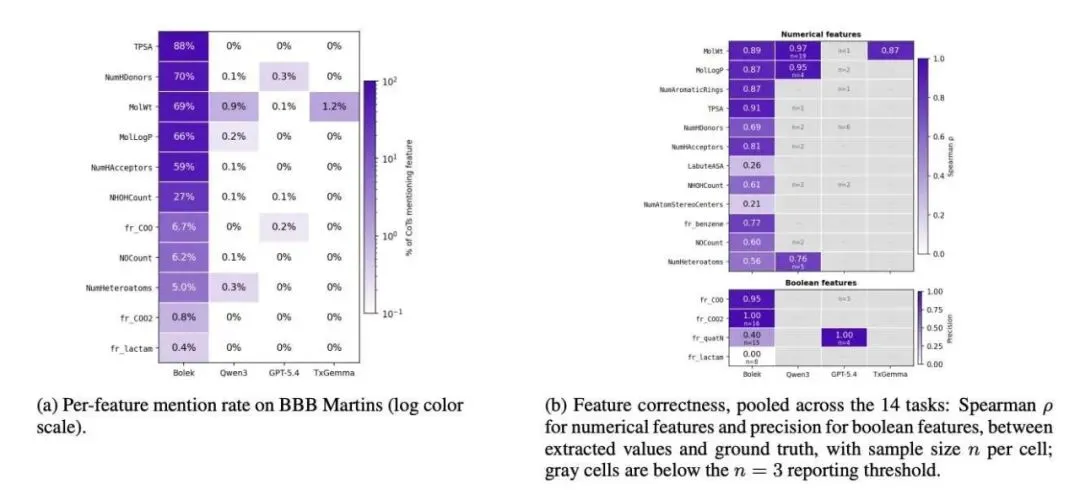
结果确实让人眼前一亮。在 15 个 TDC 任务中,BOLEK 在常规问答模式下的平均 ROC/PR AUC 从基础版的 0.55 直接跃升到了 0.76。它的参数量不到 TxGemma-9B-Chat 的一半,但在 13 个任务上都赢了对方。
作为一个药物研发人员,我最看重的是它的「接地气」程度(Groundedness)。BOLEK 在给出推理过程时,提及具体数值描述符的频率比 Qwen3、TxGemma 甚至 GPT 都要高出 10 到 100 倍。它写出的 TPSA 或 LogP 数值,跟 RDKit 算出来的真实值高度相关(Spearman 相关系数在 0.87 到 0.91 之间)。你可以拿着计算器去核对它的推理过程,这种可审计的推理在 AI 制药界是稀缺品。
这篇论文还做了一个有意思的消融实验,对比指纹和 SMILES 到底哪个好用。结论是各有千秋。在 CYP 酶或 P-gp 外排转运蛋白这些高度依赖局部子结构和三维形状的任务上,指纹完胜。在需要全局线索或存在多重机制的任务(比如 HIV)上,SMILES 表现更好。这也印证了行内人的直觉:在药物发现中,没有哪一种分子表征可以搞定所有问题。
对于未见过的任务,BOLEK 也能做到不错的泛化。在零样本(Zero-shot)条件下,它不仅在多个未参与训练的分类任务上表现优异,甚至在脂溶性或溶解度等纯回归任务上依然展现出了有意义的排序相关性。
📜Title: BOLEK: A Multimodal Language Model for Molecular Reasoning
🌐Paper: https://arxiv.org/abs/2605.02745

3. AI Agent 团队上超算,能搞定「不可成药」蛋白吗?
在药物研发领域,本质无序蛋白(Intrinsically Disordered Proteins, IDP)一直是一块难啃的硬骨头。这些蛋白没有固定的三维结构,像一团煮烂的面条,让基于结构的药物设计方法无从下手。很多 AI 公司宣称能设计蛋白,但如果你给它一个没有稳定口袋的 IDP 靶点,大多数模型就只能干瞪眼。
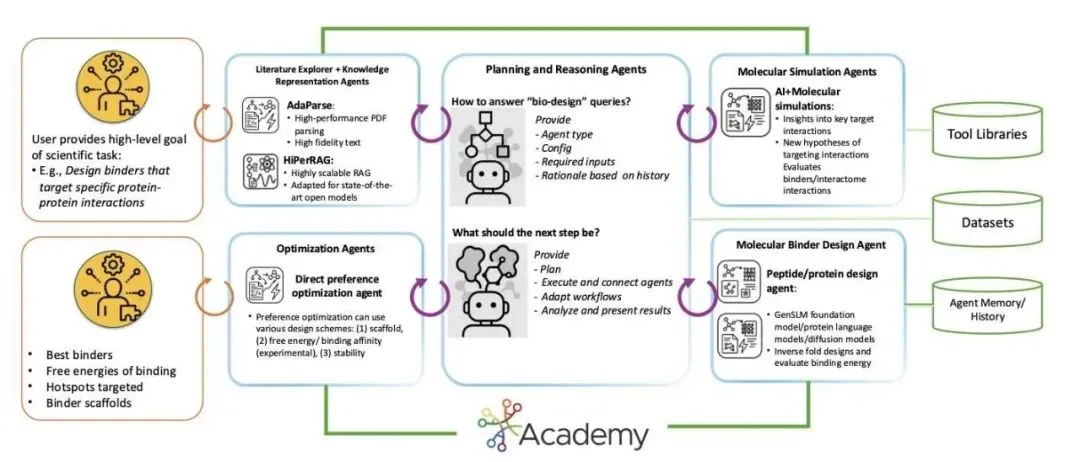
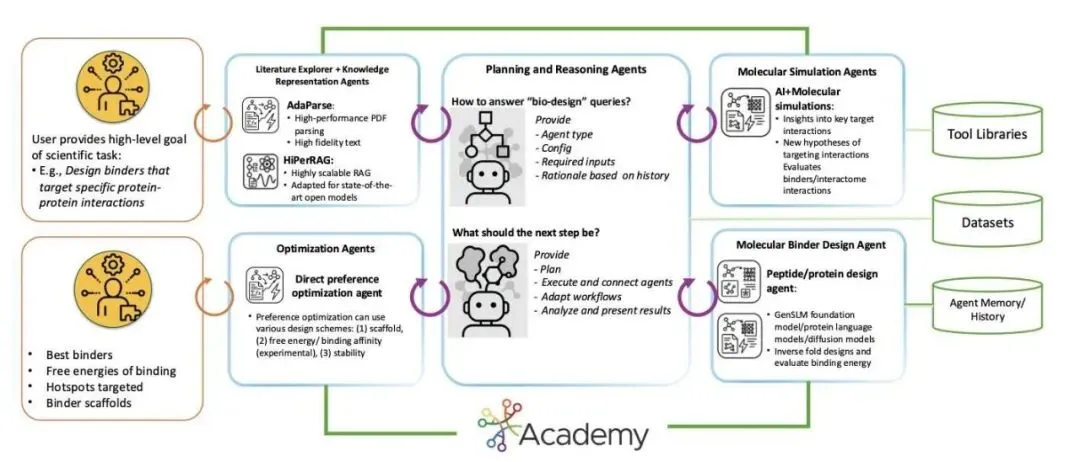
最近一个叫StructBioReasoner的系统,思路完全不一样。他们没有去训练一个更大的生成模型,而是构建了一个由多个 AI「智能体」(Agent)组成的自动化科研团队,让它在超级计算机上自主地解决「下一步该设计什么」这个问题。
工作原理:一场计算化学的「锦标赛」
想象一下,这个系统不是一个黑箱,而是一个分工明确的团队。
「假设」智能体:它会先提出一些想法,比如「我认为这个 IDP 的这段无序区域可能是一个关键的结合位点」,或者「我们可以试试用这种类型的蛋白骨架去结合它」。 「执行」智能体:拿到这些假设后,它立刻开始干活。它会调用各种计算工具,比如用结构预测模型(如 Chai-1、Boltz-2x)生成初始的三维结构,然后用分子动力学(Molecular Dynamics, MD)模拟来观察设计的分子是否真的能稳定地结合在靶点上。 「裁判」智能体:它负责评估模拟结果。它会计算结合自由能(MM-PBSA)、分析稳定性(RMSD/RMSF)等一系列指标,判断哪些设计是「好」的,哪些是「坏」的。
这整个过程就像一场锦标赛。每一轮,表现最好的设计方案会被「晋级」,为下一轮的设计提供思路。而那些失败的方案也不会被白白扔掉,系统会给它们打上「失败原因」的标签,让后续的设计能吸取教训。这种循环往复、不断迭代优化的方式,比一次性生成几千个分子然后筛选要智能得多。
专为 IDP 打造的「屠龙技」
这套系统最妙的地方在于它如何处理 IDP 的动态性。
既然 IDP 本身没有稳定结构,那就看它在细胞里都和谁「玩」。系统内置了一个名为HiPerRAG的文献检索模块,它能吞下与靶点相关的成千上万篇论文,从中提取出这个 IDP 的相互作用网络(interactome)。
有了这张「社交关系图」,系统就能知道 IDP 在与不同伙伴结合时会呈现出哪些特定的、有功能的构象。它的目标不再是凭空设计一个能结合「乱麻」的分子,而是设计一个能模仿其天然伴侣、并阻断某个关键相互作用的生物药。这相当于把一个「不可成药」的难题,转化成了一个有生物学依据、可以计算求解的问题。
结果如何?实践是检验真理的唯一标准
研究者们用两个案例检验了这套系统。
首先是一个相对简单的、有固定结构的过敏原蛋白Der f 21。这是一个「新手村」任务,用来验证系统本身是否靠谱。结果,系统设计的 50.98% 的分子,其预测结合能力都超过了文献中由人类专家设计的参照物。这说明,基本功是扎实的。
接下来是真正的挑战——富含 IDP 区域的蛋白NMNAT-2。系统首先通过「阅读文献」确定了它的 18 个相互作用伙伴,然后针对这些相互作用界面展开了大规模设计。总共生成了 266,606 个候选分子,其中 97,066 个通过了初步的结构筛选,并进入了昂贵的显式溶剂分子动力学模拟。
分析发现,84.5% 的成功设计都结合到了 IDP 区域,并且呈现出三种主要的结合模式。这证明了系统确实能识别并有效靶向 IDP 的功能界面,其结果与已知的生物学机制高度吻合。
这对我们意味着什么?
这篇工作更像是一个关于「如何做药」的方法论蓝图,而不是一个具体的药物发现。它展示了未来药物设计的一种可能性:由 AI 智能体驱动,在超级计算机上运行一个端到端的、自动化的发现引擎。
当然,这套系统的门槛极高。从他们在百亿亿次级超算 Aurora 上的扩展性测试就能看出,运行这种规模的计算需要巨大的资源。比如,在超过 64 个计算节点后,MM-PBSA 计算就遇到了文件 I/O 瓶颈。这提醒我们,再强大的算法也需要扎实的工程实现来支撑。
这套系统代表了药物发现从「AI 辅助」到「AI 自主」的又一次尝试。它不再是一个单纯的工具,而是一个能自主规划、执行和学习的「虚拟科学家」。
📜Title: Scalable Agentic Reasoning for Designing Biologics Targeting Intrinsically Disordered Proteins
🌐Paper: https://arxiv.org/abs/2512.15930
💻Code: https://github.com/IDeA-ANL-ORNL/StructBioReasoner/
4. 序列比对洗牌:Ankh 击败 AlphaFold3
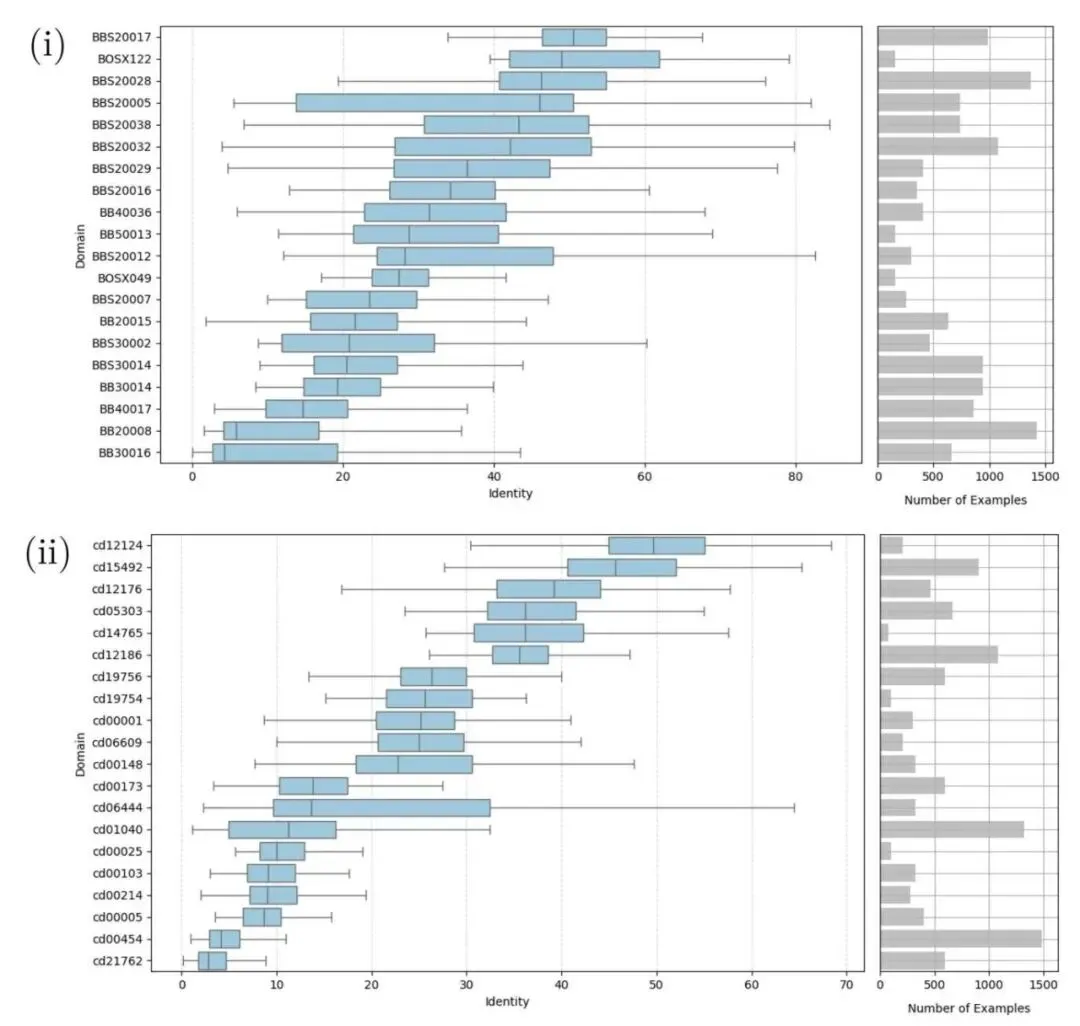
很长一段时间里,我们在做双序列比对时,脑子里只有两套范式。
老办法是用 BLOSUM 替换矩阵,这东西从 90 年代用到现在,稳定但略显陈旧。新办法是跟着 AlphaFold 带来的认知升级:既然在进化过程中,蛋白质的结构比序列更保守,那我们理应先用 AI 预测出结构,把三维结构对齐,再把对应关系反推回序列上。这听起来天衣无缝,也是目前业内很多计算生物学家推崇的路线。
但最近的一项研究打破了这个固有印象。研究者把三种比对方法放在一起硬碰硬:经典的 BLOSUM 矩阵、基于 AlphaFold3 预测再用 US-align 做结构比对的方法(AF3US),以及直接用大语言模型 Ankh 提取特征来打分的新方法(Ankh-score)。
在 BAliBASE 和 CDD 这两个经典数据集上,Ankh-score 赢了。
这是它的工作原理:研究者没有去动那些底层框架,依然沿用带有仿射空位惩罚的经典动态规划算法。他们只改了一个地方——把静态的氨基酸替换得分,换成了 Ankh 模型提取的残基级上下文特征的余弦相似度。这实际上是把蛋白质大语言模型(PLM)变成了一个即插即用的全局打分函数。
AlphaFold3 这条路线是怎么走的呢?先让 AlphaFold3 预测出两个蛋白质的结构,然后用 US-align(研究者测试后认为它在这里比 DALI 和 Foldseek 更好用)把结构对齐,最后把残基的对应关系映射回序列。
评估做得极度扎实。研究者对每个结构域的多序列比对(MSA)数据集进行了全排列的双序列比对,用了四个不同的距离指标来衡量误差,并做了严格的统计学检验。
数据揭示了一个很有意思的现象。无论序列一致性高低,还是结构相似度(TM-score)如何分层,Ankh-score 都占据统治地位。AF3US 虽然在结构相似度较高(TM-score > 0.5)的区间里赢了老旧的 BLOSUM45,但在这个它本该擅长的「结构友好」区,依然输给了 Ankh-score。
为什么 AlphaFold3 会输?
看看那些失败案例就能明白症结所在。结构比对在处理特定类型的蛋白质时会「迷路」。比如遇到含有重复序列的蛋白质,AF3US 经常把错误的 WH2 基序对齐在一起;在处理多结构域蛋白时,它甚至会找错 SH2 结构域,或者直接漏掉第二个结构域。哪怕这时候系统给出的整体 TM-score 看着挺高,生物学意义上的比对已经完全乱套了。更有趣的是,即便是在序列高度相似的情况下,AF3US 依然会出现非零误差的「动摇」。这说明结构匹配的失败不能单纯归结为 TM-score 偏低,三维空间的对齐有时无法还原真实的进化映射。
相比之下,语言模型看过海量的序列演化历史。Ankh-score 在这些容易错配的例子中给出了与人工校对近乎一致的答案。
这项研究还顺带测试了其他的蛋白质大语言模型,比如 ProstT5、ProtT5 和 ESM-C。它们用来做比对打分时,表现全都超越了 AF3US。这证明了一个核心逻辑:大语言模型的特征空间里,藏着对齐序列所需的关键进化信号,而这些信号并没有被 AlphaFold3 预测出的静态三维结构完全捕捉。
文章最后提到了一个微小但好玩的对照实验。在少数人工精选的样本上,基于 AlphaFold3 预测结构得出的比对结果,胜率竟然微微高于基于真实实验结构的比对结果。这提示我们需要重新审视现有的比对黄金标准和参考数据集里的瑕疵。
这项工作确立了 Ankh-score 在当前双序列比对领域的顶尖地位。它也给从业者提了个醒:只要你使用的「序列」是由现代大语言模型提取的动态上下文特征,那「结构比对将取代序列比对」的论断就需要重新改写了。
📜Title: E-score: Ankh-Score Produces Better Sequence Alignments Than AlphaFold3
🌐Paper: https://doi.org/10.1002/prot.70143
💻Code: http://github.com/lucian-ilie/E-score
5. DoFormer 预测基因扰动:告别 DAG 图假设
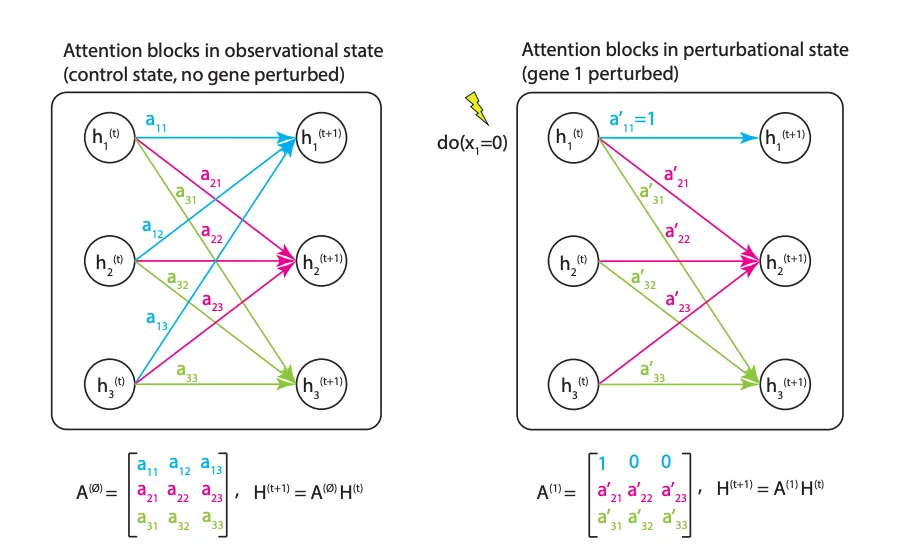
当做药物靶点发现时,最核心的问题永远是:「如果我们敲除这个基因,或者过表达那个基因,细胞会发生什么变化?」
现在大家都在用 Perturb-seq(CRISPR 结合单细胞测序)来做高通量筛选。穷举所有基因和所有细胞类型的组合是不现实的,成本太高。我们需要计算模型来预测那些还没做过实验的扰动结果。
一直以来,因果发现算法都面临一个巨大的硬伤:它们极度依赖有向无环图(DAG)假设。这种假设规定因果关系只能单向流动,不能形成闭环。稍微懂点生物学的人都知道这有多离谱。基因调控网络里到处都是反馈回路。基因 A 激活 B,B 激活 C,C 回过头来抑制 A。DAG 算法在处理这种真实生物网络时往往会失效。
这就是 DoFormer 这项研究吸引我的地方。研究者找到了一种绕过 DAG 假设的巧妙方法。
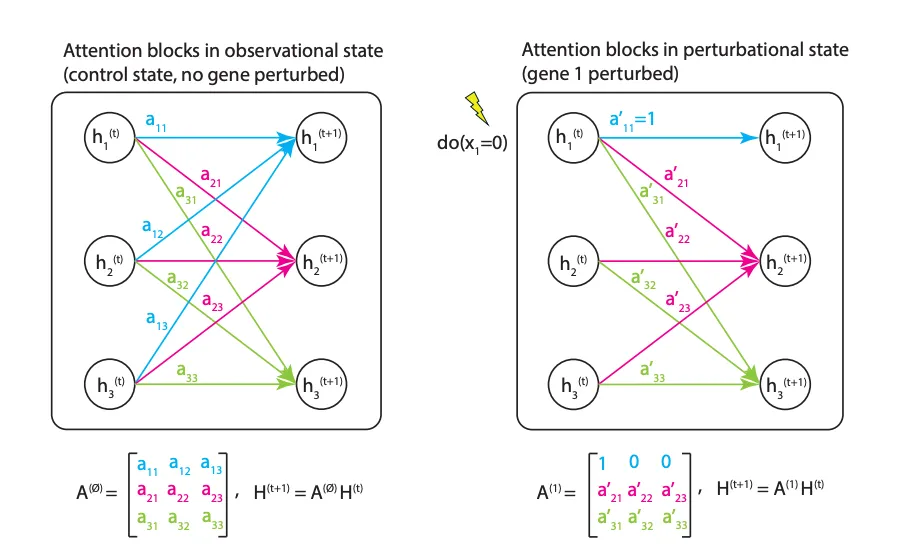
这是它的工作原理。DoFormer 直接把 Judea Pearl 著名的因果推断理论「do-算子」硬编码到了 Transformer 的注意力机制里。设想我们要对基因 P 施加干预(比如 CRISPR 敲除)。模型会做两件事。第一,把基因 P 的输入表达值强行设定为干预值(敲除就是 0)。第二,它直接把注意力矩阵里基因 P 对应的那一行清零。
这步操作非常符合直觉。当你人为敲除一个基因时,它就不再受上游调控信号的影响了。把注意力矩阵清零,相当于在算法层面切断了这个基因接收其他基因信号的通路。它保留了对下游基因的影响力,完美模拟了真实的生物学干预过程。这样做完全不需要什么 DAG 假设,自然也就容许了反馈回路的存在。
为了让预测更准,模型引入了两个层面的数据。它不仅看单细胞特异性的 RNA 表达量,还引入了蛋白质语言模型(PLM,如 ESM2)提取的蛋白质序列特征。RNA 告诉你细胞现在正在发生什么,而蛋白质序列为模型提供了一个不受细胞状态影响的物理学基线。这两个特征被线性投影并拼接在一起,构成了每个基因的底层表示。
训练过程分为两步。先做预训练。研究者拿对照组细胞的数据,随机遮盖掉 15% 的基因表达值,让模型去猜。模型搞清楚了基因之间基本的共表达相关性。
然后进入微调阶段。这一步要让模型从「相关」走向「因果」。他们用真实的扰动数据进行训练,采用了一种加权均方误差(WMSE)损失函数。权重来自于差异表达(DE)的统计学得分。如果一个基因在真实实验中发生了显著的表达差异,模型在训练时就必须给予它更多的关注。
效果如何?研究者在一个包含超过 60 万个细胞、跨越 4 种细胞类型的庞大 Perturb-seq 数据集上进行了测试。结果显示,DoFormer 的表现明显优于此前的 STATE 模型和常规基准。特别是在预测那些发生了显著差异表达的基因时,它的准确度提升极大。
随着目标细胞类型中可用的训练数据越来越多,DoFormer 的性能会稳步上升,表现出了很好的因果一致性。即便面对「零样本」挑战——也就是某个基因的扰动数据在训练集中从未出现过——它依然能给出靠谱的预测方向。
从消融实验来看,模型参数从 170 万扩大到 2700 万,整体预测能力获得了全面提升。虽然大模型对蛋白质序列特征的依赖度有所降低,但在默认情况下加入这些多模态信息依然能带来稳健的收益。
利用注意力机制重塑因果干预,这是一个极具启发性的思路。它让 AI 真正学会了用干预者的视角来审视基因网络。
📜Title: DoFormer: Causal Transformer for Gene Perturbation
🌐Paper: https://www.biorxiv.org/content/10.64898/2026.05.02.722054

