 夜雨聆风
夜雨聆风

作者
Eric A Stach(宾夕法尼亚大学)
Ping Liu(纽约州立大学石溪分校)
谢建(普渡大学)
引言
质子交换膜燃料电池(PEMFCs)阴极氧还原反应(ORR)的缓慢动力学是制约其商业化的关键瓶颈。Pt基催化剂作为ORR基准催化剂,存在成本高、储量有限及运行条件下性能衰减等问题。Pt-M合金(如Pt-Co、Pt-Ni等)通过引入压缩晶格应变和电子效应可减弱Pt-OH键强度,提升ORR活性,但常因结构不稳定和过度晶格压缩导致性能下降。密度泛函理论(DFT)研究表明,当*OH结合能比纯Pt(111)弱0.10-0.12 eV时可达到ORR活性火山图峰值,对应-1.0%至-1.4%的压缩应变。稀土(RE)元素合金化可显著提升结构稳定性,并通过镧系收缩效应精确调控晶格应变。本研究通过DFT计算确定Pt5Co亚层可诱导最优压缩应变(-1.24%),并利用Ce元素作为结构模板稳定热力学不稳定的Pt5Co相,成功制备出兼具高活性和优异耐久性的Pt5(Ce)Co@Pt多层纳米催化剂。
核心发现
本研究通过理论计算与实验验证相结合的策略,成功设计并合成了Pt5(Ce)Co@Pt多层纳米催化剂。DFT计算表明,Pt5Co(111)亚层可诱导-1.24%的最优压缩应变,使*OH结合能偏移0.107 eV,恰好位于ORR活性火山图峰值区域。通过稀土Ce模板策略稳定了热力学不稳定的Pt5Co相,形成了Ce富集核心-Pt5Co类亚层-Pt富集壳层的多层结构。电化学测试显示,该催化剂在RDE测试中质量活性达2.6 A·mgPt⁻¹,半波电位0.93 V,显著优于Pt3Co@Pt和PtCo@Pt。在MEA测试中,重型车条件下电流密度达1.88 A·cm⁻²(0.7 V),经180,000次AST循环后仍保持1.17 A·cm⁻²,远超DOE 2025目标(90,000次循环后1.07 A·cm⁻²)。结构表征证实Ce模板不仅实现了Pt5Co类结构的可控制备,还有效抑制了原子扩散和结构坍塌,使催化剂在长期运行中保持优异稳定性。该工作展示了理论指导应变工程与稀土模板相结合的设计策略在开发高性能燃料电池催化剂方面的巨大潜力。
图文解读
图1:DFT计算指导的应变-活性关系与火山图预测
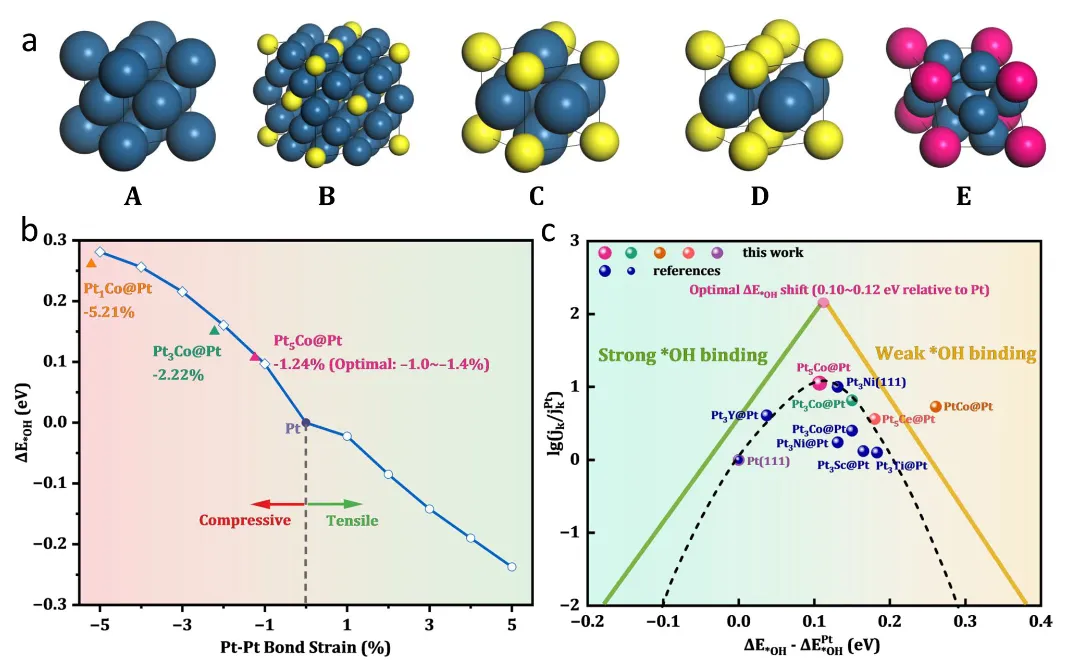
图1展示了DFT计算指导催化剂设计的理论基础。图1a呈现了Pt、Pt5Co、Pt3Co、Pt1Co和Pt5Ce的体相晶格结构,通过调节Pt:Co比例可系统调控晶格参数。图1b揭示了Pt-Pt键应变与*OH结合能偏移(ΔE*OH)的线性关系,随着压缩应变增加(从Pt5Co的-1.24%到PtCo的-5.21%),*OH结合能逐渐减弱,体现了应变效应对表面吸附性质的主导作用。图1c为经典的ORR活性火山图,横轴为ΔE*OH,纵轴为质量活性,峰值区域对应ΔE*OH约0.10-0.12 eV。Pt5Co@Pt的计算数据点恰好落在火山图峰值附近,预测其具有最优ORR活性,而Pt3Co@Pt和PtCo@Pt因过度压缩应变偏离最优区域。该图还标注了文献报道的Pt(111)、Pt3Y@Pt、Pt3Ni(111)等参考数据点,验证了理论模型的可靠性。这一理论预测为后续实验合成提供了明确的组分与结构指导,即通过调控Co含量实现约-1.24%的最优压缩应变。
图2:Pt5(Ce)Co@Pt多层纳米结构的合成与表征
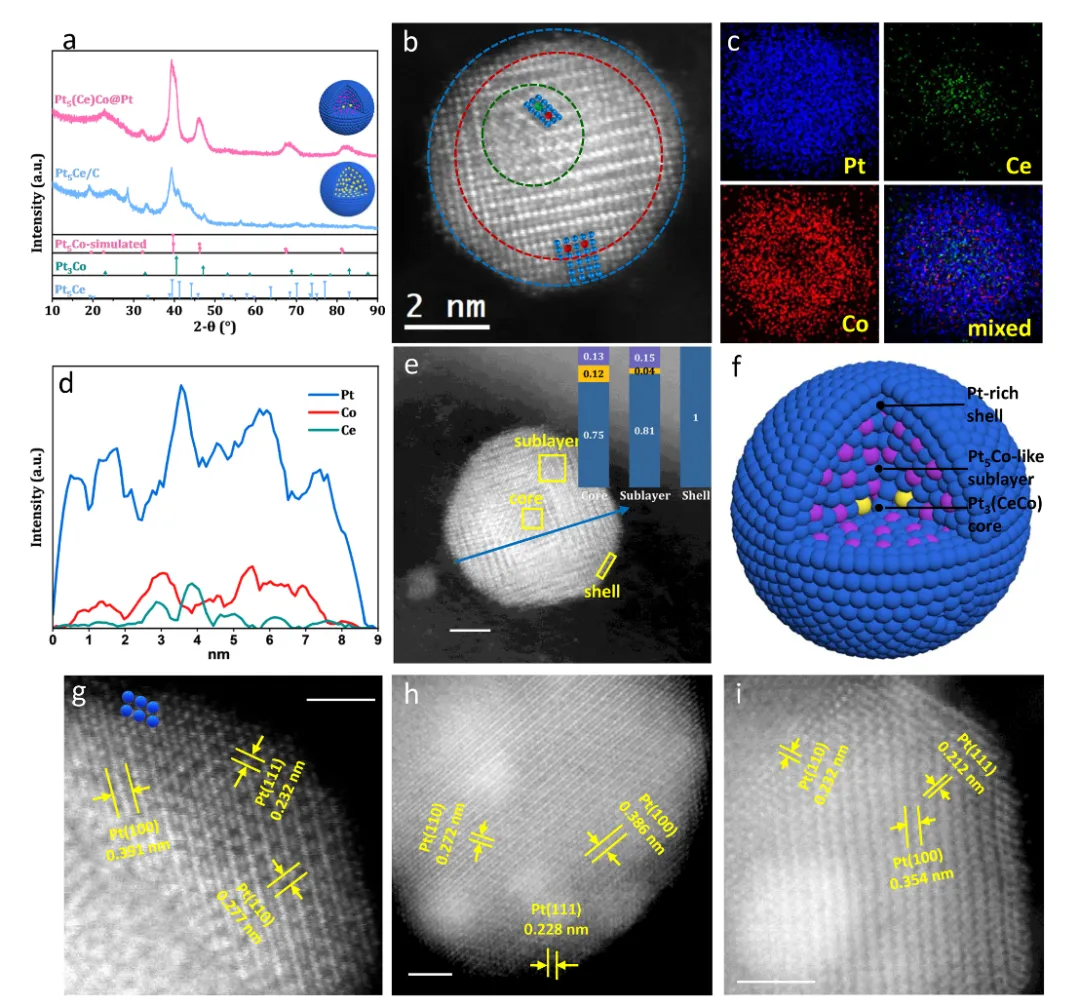
图2详细展示了Pt5(Ce)Co@Pt催化剂的结构特征与形成机制。图2a的XRD图谱显示,前驱体Pt5Ce呈现典型的CaCu5型六方相特征峰(PDF#17-0071),而经Co扩散和酸刻蚀后的Pt5(Ce)Co@Pt转变为L12型面心立方衍生结构,主峰位置与模拟的L12-Pt5Co图谱吻合,证实了从六方相到立方相的结构转变。Rietveld精修结果表明样品包含68.8 wt.%的Pt3Co类相、19.2 wt.%的Pt5Ce类相和12 wt.%的金属Pt,反映了Co掺杂后的多相混合特征。图2b的HAADF-STEM图像清晰揭示了纳米颗粒的多层结构:中心区域(绿色虚线圈)显示平行排列的原子柱,亮度变化表明部分有序化;向外延伸的中间层呈现更规则的原子排列,对应Pt5Co类亚层;最外层为低对比度的Pt富集壳层。这种独特的Ce核心-Pt5Co亚层-Pt壳层的三层结构正是Ce模板策略的成功体现。图2c的EDS元素分布图进一步证实了结构特征:Ce元素集中在颗粒核心,Co元素均匀分布在亚层区域,Pt元素覆盖整个颗粒表面,形成了清晰的元素梯度分布。线扫描剖面定量展示了从中心到表面的元素浓度变化,Ce信号在核心区域最强,Co信号在亚层区域达到峰值,Pt信号在壳层区域占主导。这一多层结构的形成机制在于:高温退火过程中,Co优先取代Pt5Ce亚表面的Ce原子(ΔE = -2.58 eV/atom),而核心区域的Ce因动力学限制和热力学稳定性得以保留,从而实现了热力学不稳定Pt5Co相的稳定化。
图3:电化学性能测试与应变效应实验验证
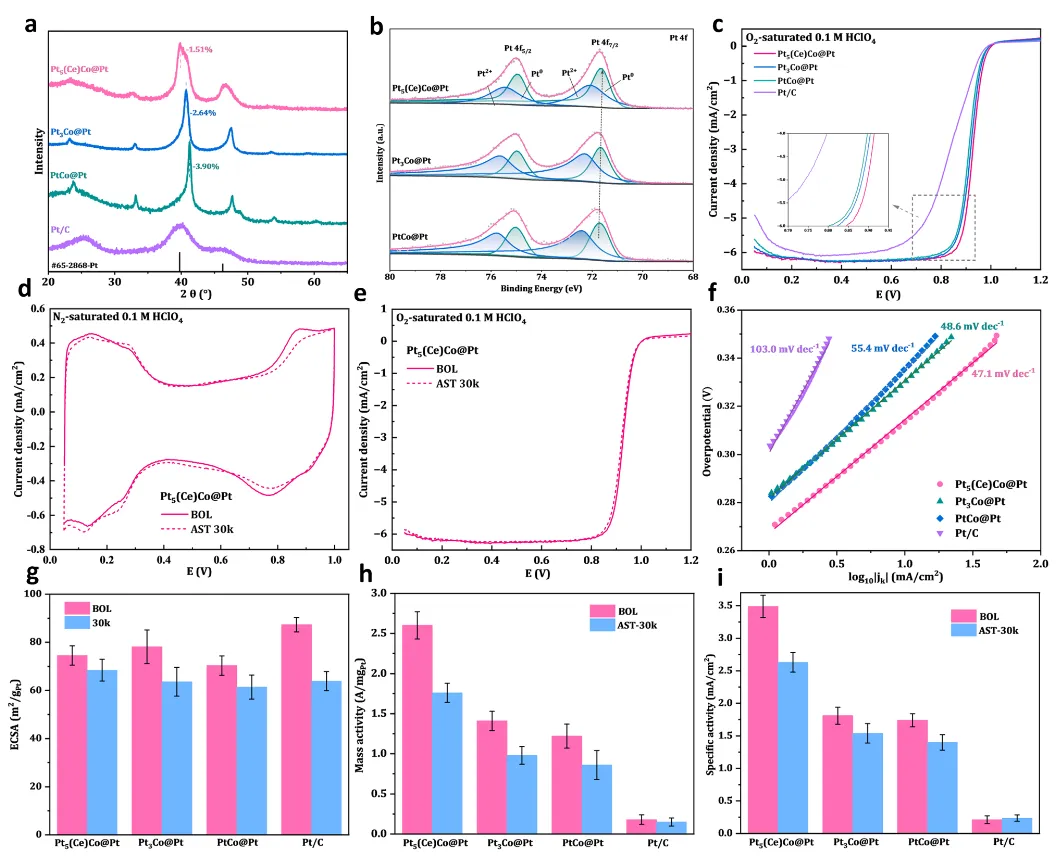
图3系统评估了催化剂的ORR性能并验证了DFT预测的应变-活性关系。图3a的XRD图谱对比了Pt5(Ce)Co@Pt、Pt3Co@Pt、PtCo@Pt和商业Pt/C的晶体结构,Pt5(Ce)Co@Pt的(111)峰位于71.8°,介于Pt3Co@Pt(71.9°)和PtCo@Pt(72.1°)之间,表明其晶格参数和应变程度适中。根据Bragg方程计算,Pt5(Ce)Co@Pt的晶格常数为3.89 Å,对应约-1.2%的压缩应变,与DFT预测的-1.24%高度吻合。图3b的XPS Pt 4f谱图显示,Pt5(Ce)Co@Pt的Pt0 4f7/2结合能为71.55 eV,低于Pt3Co@Pt(71.68 eV)和PtCo@Pt(71.91 eV),反映了压缩应变导致的Pt d带中心下移,与理论预测的电子结构变化一致。Pt5(Ce)Co@Pt的Pt0/Pt2+峰面积比(1.54)最高,表明表面金属度高、氧化物少,有利于催化活性位点暴露。图3c的ORR极化曲线显示,Pt5(Ce)Co@Pt在0.9 V下的质量活性达2.6 A·mgPt⁻¹,分别是Pt3Co@Pt(1.41 A·mgPt⁻¹)和PtCo@Pt(1.22 A·mgPt⁻¹)的1.8倍和2.1倍,远超商业Pt/C(0.18 A·mgPt⁻¹)。半波电位(E1/2)依次为0.93 V(Pt5(Ce)Co@Pt)、0.92 V(Pt3Co@Pt)、0.91 V(PtCo@Pt),正移趋势与应变优化程度一致。图3f的Tafel斜率分析进一步证实了动力学优势,Pt5(Ce)Co@Pt的Tafel斜率最低(47.1 mV·dec⁻¹),表明ORR反应能垒最小,与最优*OH结合能的理论预测相符。图3d,e展示了30,000次AST循环前后的CV和ORR曲线,Pt5(Ce)Co@Pt质量活性保留率达69%(2.6降至1.8 A·mgPt⁻¹),ECSA损失仅8%,表现出优异的稳定性。图3g-i量化了活性衰减程度,Pt5(Ce)Co@Pt的质量活性损失(31%)与Pt3Co@Pt(30%)和PtCo@Pt(29%)相当,但其初始活性显著更高,体现了Ce模板对结构稳定性的增强作用。这些电化学结果完美验证了DFT预测的应变-活性关系,证实Pt5(Ce)Co@Pt通过最优压缩应变实现了火山图峰值活性。
图4:膜电极组装(MEA)测试与重型车耐久性验证
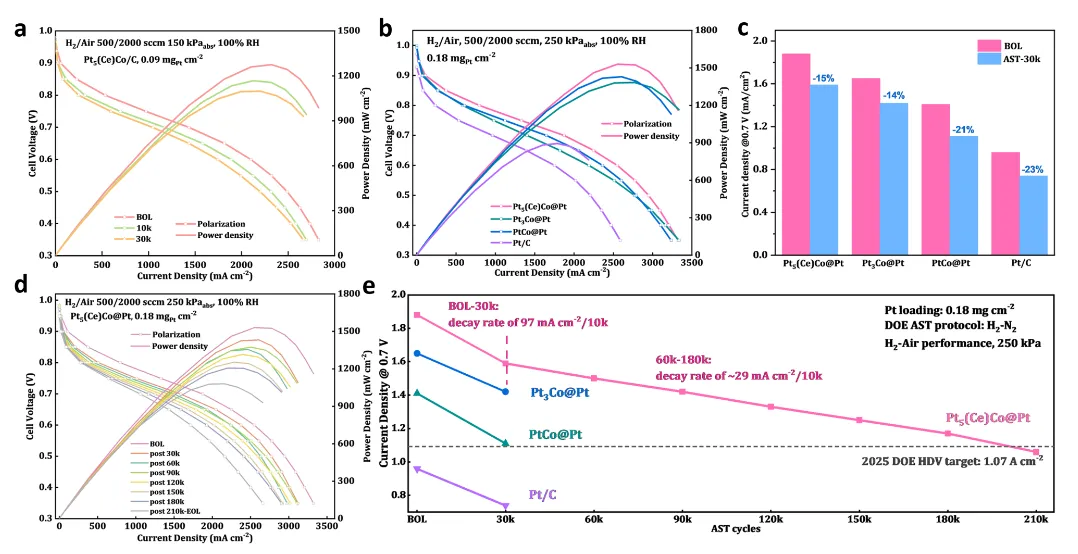
图4展示了催化剂在实际燃料电池器件中的性能表现。图4a为轻型车(LDV)条件下的H2/空气极化曲线,Pt5(Ce)Co@Pt在0.8 V下达0.59 A·cm⁻²,0.9 V下的质量活性为1.58 A·mgPt⁻¹,远超DOE 2025目标(0.3 A·cm⁻² at 0.8 V, 0.44 A·mgPt⁻¹ at 0.9 V)。经10,000和30,000次AST循环后,电流密度仅轻微下降,展现了良好的运行稳定性。图4b对比了重型车(HDV)条件下各催化剂的初始性能,Pt5(Ce)Co@Pt在0.7 V下电流密度达1.88 A·cm⁻²,质量活性0.78 A·mgPt⁻¹(0.9 V),显著高于Pt3Co@Pt(1.65 A·cm⁻², 0.68 A·mgPt⁻¹)、PtCo@Pt(1.41 A·cm⁻², 0.53 A·mgPt⁻¹)和商业Pt/C(0.96 A·cm⁻², 0.12 A·mgPt⁻¹),证实了RDE测试中的高活性可成功转化为全电池性能。图4c统计了30,000次AST循环后的电流密度损失,Pt5(Ce)Co@Pt从1.88降至1.59 A·cm⁻²(15%损失),与Pt3Co@Pt(14%损失)和PtCo@Pt(21%损失)相当,表明多层结构未牺牲稳定性。图4d,e展示了长期耐久性测试结果,这是本研究的核心亮点。在HDV条件下经180,000次AST循环后,Pt5(Ce)Co@Pt仍保持1.17 A·cm⁻²(0.7 V),远超DOE 2025目标要求的90,000次循环后1.07 A·cm⁻²。衰减曲线呈现两阶段特征:前30,000次循环衰减较快(15.4%损失,97 mA·cm⁻²/10,000次循环),归因于小颗粒的Ostwald熟化和表面重构;60,000次循环后进入稳态,衰减率降至29 mA·cm⁻²/10,000次循环(约2%),表明系统达到准稳态。这一优异耐久性源于Ce模板对Pt5Co亚层的结构稳定作用,抑制了原子扩散和相坍塌,使活性结构在长期运行中得以保持。该结果不仅突破了DOE目标,更展示了稀土模板策略在提升催化剂耐久性方面的巨大优势。
图5:长期循环后的结构演变与稳定性机制
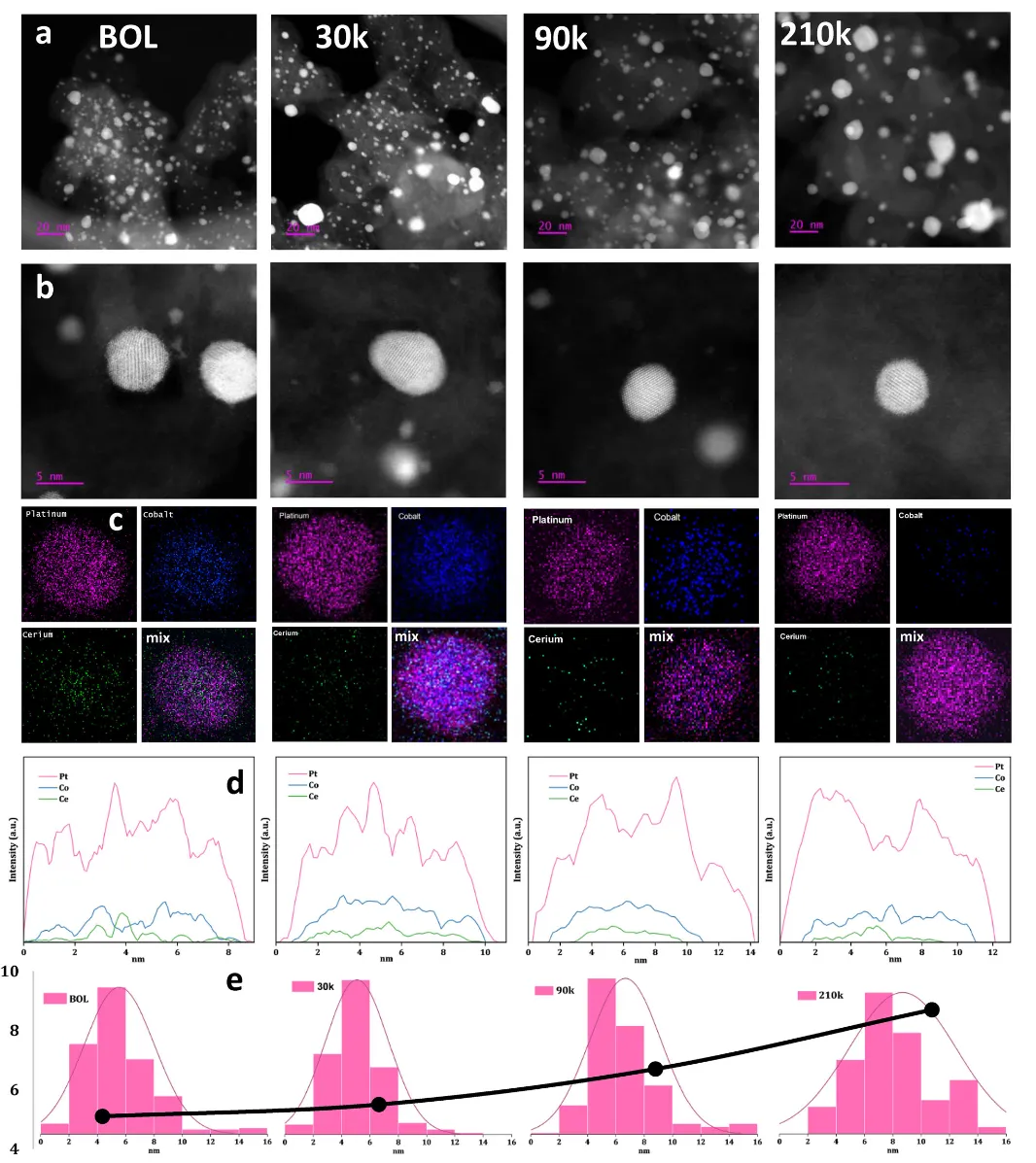
图5通过多尺度结构表征揭示了催化剂在长期AST循环后的演变机制。图5a的低倍STEM图像展示了从初始(BOL)到210,000次AST循环的颗粒形貌变化。BOL时颗粒尺寸分布窄(平均5.1 nm),经30,000次循环后分布基本不变(平均5.5 nm),表明早期结构稳定。90,000次循环后小颗粒(<4 nm)数量明显减少,平均尺寸增至6.7 nm,反映了颗粒迁移和Ostwald熟化的协同作用。210,000次循环后尺寸分布向大尺寸偏移(平均8.7 nm),2-6 nm范围颗粒显著减少,体现了持续的颗粒粗化过程。图5b的HAADF-STEM图像和图5c的EDS元素分布图揭示了多层结构的演变特征。30,000次循环后,Ce核心和Co富集亚层仍清晰可见,Pt壳层保持完整,证实了结构的早期稳定性。随着循环次数增加,Pt壳层逐渐增厚,表明运行过程中发生了表面重构和Pt再沉积。值得注意的是,即使在210,000次循环后,仍有颗粒保持核-壳结构,尽管成分对比度有所降低,这体现了Ce模板框架对结构完整性的保护作用。图5d的线扫描剖面定量展示了元素分布演变:随着循环进行,Co和Ce从外层区域逐渐流失,结合Pt表面富集,形成了稳定的配置,抑制了进一步快速衰减。这些观察结果支持了两阶段衰减机制:初始阶段(30,000次循环)结构变化有限;中间阶段(30,000-90,000次循环)小颗粒溶解或合并;后期阶段(>90,000次循环)系统达到准稳态,厚Pt壳层和保留的核-壳结构使衰减率显著降低。Ce模板架构通过增强Pt5Co类亚层的结构和热力学稳定性,减少了原子迁移,在长期运行中保持了活性结构的完整性,这是催化剂实现超长耐久性的关键机制。
总结
本研究通过DFT理论计算与实验验证相结合的策略,成功设计并制备了Pt5(Ce)Co@Pt多层纳米催化剂,实现了ORR活性与耐久性的协同优化。理论计算表明,Pt5Co(111)亚层可诱导-1.24%的最优压缩应变,使*OH结合能偏移0.107 eV,恰好位于ORR活性火山图峰值。通过稀土Ce模板策略稳定了热力学不稳定的Pt5Co相,形成了Ce富集核心-Pt5Co类亚层-Pt富集壳层的独特多层结构。该催化剂在RDE测试中质量活性达2.6 A·mgPt⁻¹,半波电位0.93 V,显著优于传统Pt-Co合金。在MEA重型车条件下,电流密度达1.88 A·cm⁻²(0.7 V),经180,000次AST循环后仍保持1.17 A·cm⁻²,远超DOE 2025目标(90,000次循环后1.07 A·cm⁻²)。结构表征证实Ce模板不仅实现了Pt5Co类结构的可控制备,还有效抑制了原子扩散和结构坍塌。该工作展示了理论指导应变工程与稀土模板相结合的设计策略在开发高性能燃料电池催化剂方面的巨大潜力,为下一代Pt基催化剂的理性设计提供了新思路。
原文链接
https://doi.org/10.1002/adma.73269
