 夜雨聆风
夜雨聆风
我有个师兄读博四年,只做了一个膜蛋白结构。 他养了一年多细胞,纯化了两年蛋白。 长晶体长到第三年,才出一颗能用的样品。 送同步辐射、解相位、优化结构,等他答辩当天,PDB编号才刚批下来。
2024年5月8日,DeepMind和同构实验室在《自然》发了论文。 论文题目是《AlphaFold 3精准预测生物分子互作结构》。 同一天,alphafoldserver.com上线。 全球研究者点几下鼠标,几分钟就能拿到蛋白复合物预测结构。 除了蛋白,DNA、RNA、小分子配体、金属离子、糖基化修饰都能算。
我师兄那篇博士论文的核心成果,现在本科生开个浏览器标签页就能复刻。
这篇文章探讨几个核心问题: AlphaFold 3到底怎么"猜"结构? 和2020年的AlphaFold 2有什么差异? 为什么这次选了扩散路径? 它真的无所不能吗?
博士生四年磨一个结构
1958年,剑桥学者John Kendrew用X射线衍射解出首个蛋白结构。 是一头鲸的肌红蛋白。 那篇《自然》论文里,肌红蛋白画成一盘盘卷起来的香肠。 Kendrew后来拿了1962年诺贝尔化学奖。 磨结构这门手艺,从此有了起点。
之后的几十年,解结构一直是苦力活。 要知道一段氨基酸序列怎么折叠,得先纯化对应蛋白、长晶体、送同步辐射,再花几个月解相位。 博士生四年做一个结构是常态。
冷冻电镜(cryo-EM) 2017年拿了诺奖。 它靠液氮急冻样品,用电子显微镜拍照重建三维结构。 膜蛋白的解析难度才降了一点,但纯化和数据处理还是很慢。 核磁共振(NMR) 靠原子周围的核磁信号反推距离约束,只适合小蛋白。
1972年学者Christian Anfinsen拿诺奖时,致辞里提了一个判断。 业内叫安芬森教条(Anfinsen dogma):生理环境下,蛋白只靠氨基酸序列就能决定三维结构,不需要额外遗传指令。 也就是说,理论上只要知道序列,结构就是唯一确定的。
但1969年学者Cyrus Levinthal提了反向提醒,业内叫莱文索尔悖论。 一条100个残基的肽链,每个二面角随机试一遍,构象空间是10^300量级。 试完的时间比宇宙年龄还长。 但真实蛋白几毫秒到几秒就能折好。 说明折叠肯定不是穷举,背后有局部规则提前压缩了搜索空间。
这两个结论一个给希望,一个泼冷水。 序列决定结构,说明可以靠计算得到。 但搜索空间太大,没人知道怎么算。 之后50年,主流研究走了两条路。
一条是David Baker团队的Rosetta软件。 2000年前后由华盛顿大学蛋白质设计研究所开发。 核心逻辑是拼小片段、用能量函数排序、蒙特卡洛算法搜全局最小值。 2010年前后,这条路能做到中等精度。
另一条是CASP蛋白结构预测竞赛。 1994年开始,每两年办一届盲测。 组委会选一批刚解出但未公开的实验结构,让全球算法盲算再公开打分。 2018年CASP13大赛,DeepMind第一次带AlphaFold 1参赛。 最难类目中位GDT_TS得分约58.9,比第二名高不少,但还达不到可用精度。
2020年11月CASP14大赛,AlphaFold 2把中位GDT_TS拉到92.4分。 组委会主席John Moult闭幕时说:这个问题在这里就算解决了。 88个最难的自由建模域中,AF2在87个上达到可用精度。 做了三十年结构的资深学者群当晚一片沉默,不少实验室要重新调整研究方向。
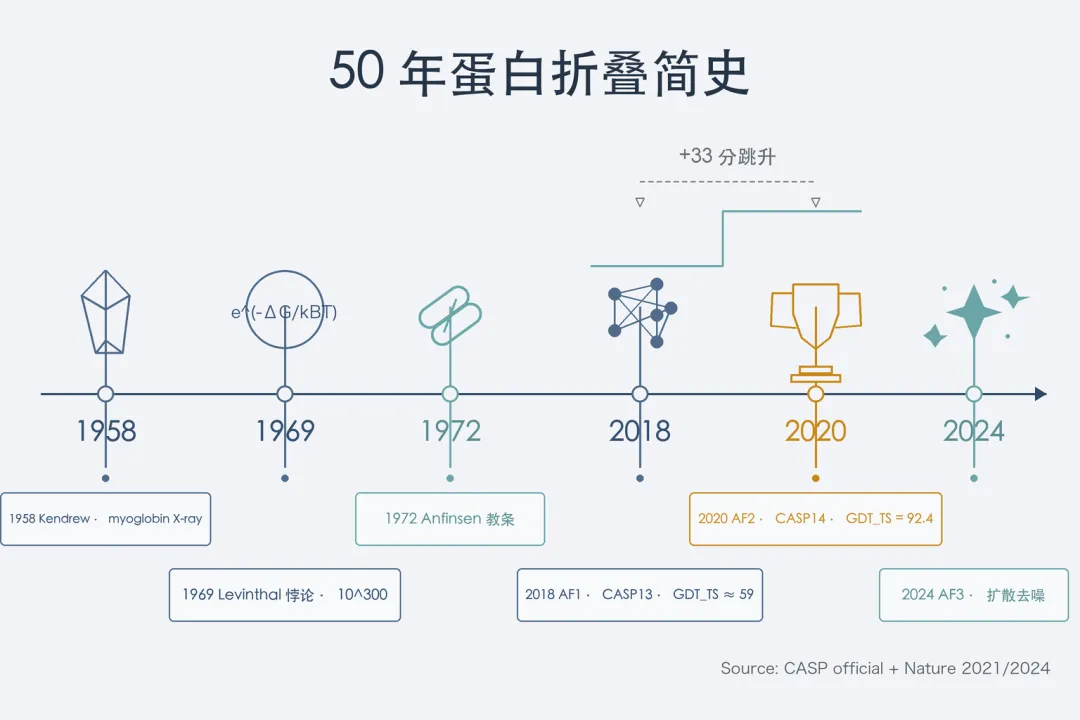
AF2把精度拉到92.4分
AF2 2021年7月正式发表,权重当天就开源。 要懂AF3的改动,得先弄清AF2的技术路径。
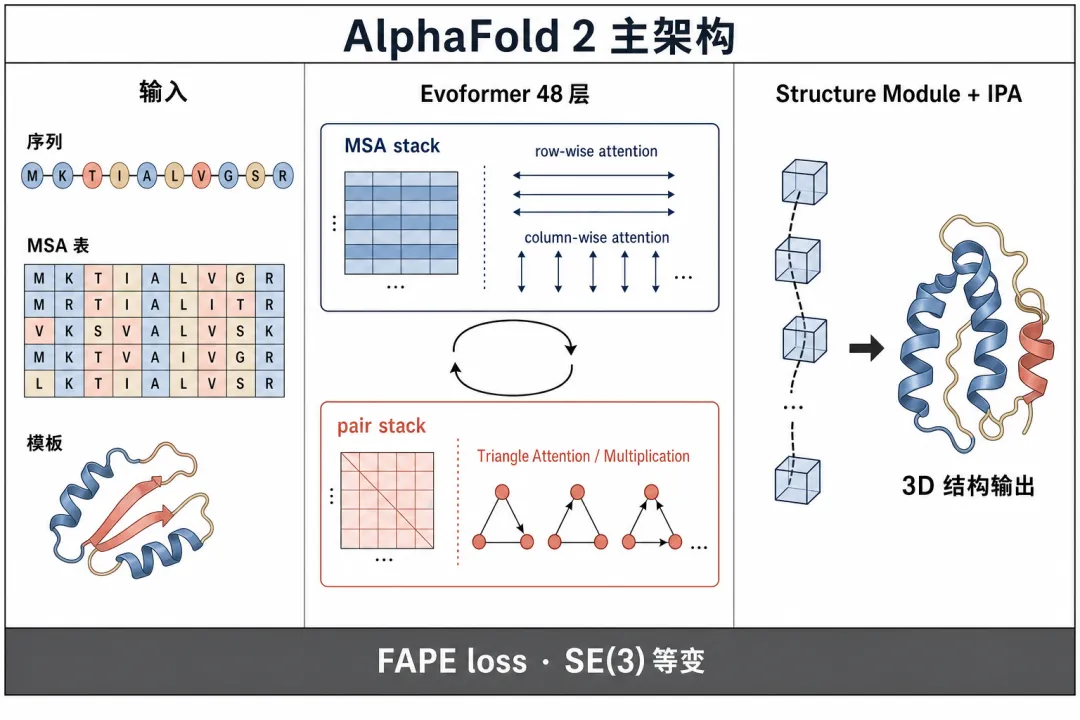
AF2的输入是一段氨基酸序列。 它不会直接画结构,先去UniRef这类几亿条的蛋白序列库,搜出几百到几千条同源序列,拼成一张表,叫多序列比对(MSA)。 就是把同源蛋白对齐排成一列,看哪些位置保守,哪些位置在不同物种里同步突变。
为什么要做多序列比对? 两个氨基酸如果在三维结构里挨着,进化压力会让它们同步突变。 一边变了,另一边为了适配也要变。 这种共变信号就埋在MSA里。 AF2之前的trRosetta、AlphaFold 1,都是从MSA里挖共变信号。
AF2的主干叫Evoformer,是48层注意力模块。 它反复更新两类信息:一类是MSA表征,行是同源序列,列是位置。 另一类是配对表征,存残基i和残基j之间的距离、角度信息。 两类信息轮流传消息,配对表征里加入三角注意力、三角乘法等几何约束,最后提炼出两两残基之间的几何先验。
之后接结构模块不变点注意力(IPA)。 这是一种在3D点云上做的几何注意力,旋转或平移整个分子,结果不变。 IPA把每个残基放进局部坐标系,反复用注意力互作,直接输出每个原子的3D坐标。
训练时用FAPE损失。 这是一种坐标系无关的位置损失,把预测原子放到真实参考坐标系里逐个比距离。 整套系统端到端可微,满足SE(3)等变性。 网络结构本身就能保证,输入旋转,输出也会跟着旋转,不用靠数据增强教模型。
这套架构非常工整,但有两个限制,让AF2只能算蛋白。 第一,IPA专为20种标准氨基酸的几何骨架设计。 要扩展到DNA、RNA、小分子,得为每种新分子重写一套几何处理逻辑。 第二,FAPE的几何先验把注意力锁死在残基刚体层级,要推到原子级精度就会受限。
后来DeepMind推出AlphaFold-Multimer处理蛋白-蛋白复合物,还有内部版本AlphaFold-Latest能跑配体。 但要做到一个模型搞定全部生物分子,AF2的几何骨架已经改不动了。
AF3重写了整条流水线
2024年5月上线的AF3,从输入到输出整条流水线全部重写。 最核心的改动有两个。
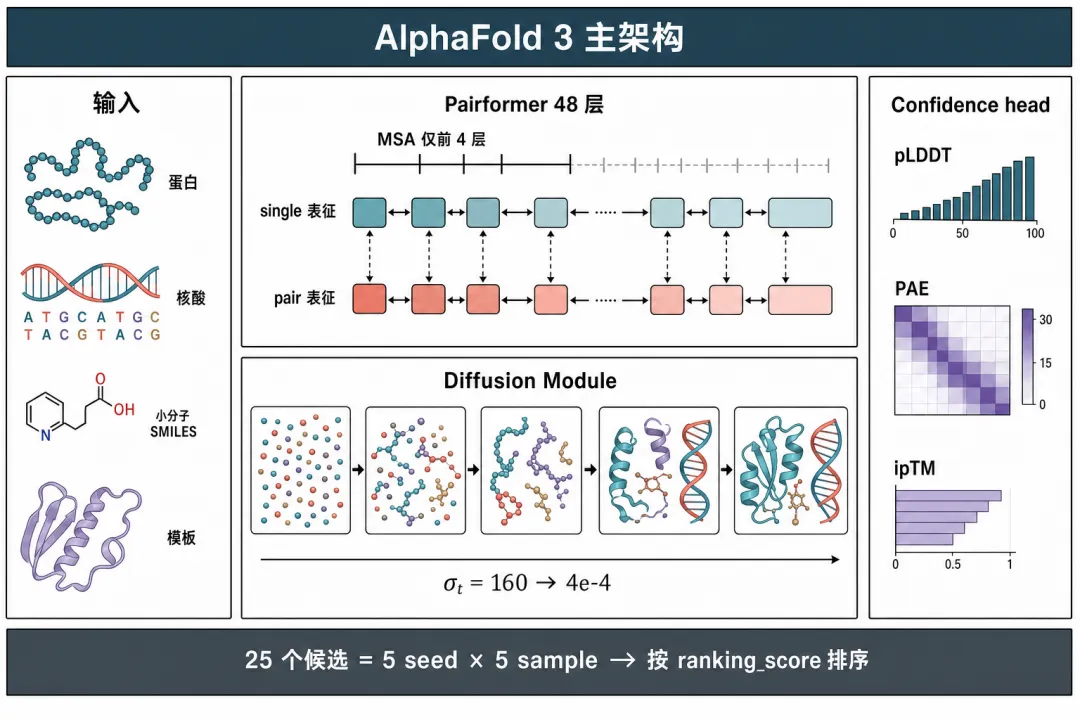
第一个改动:Evoformer缩成Pairformer。 AF3把MSA处理模块从48层砍到4层。 MSA只在最开始过一遍,把共变信号提炼到配对表征上就退出。 剩下48层叫Pairformer,只处理配对表征和单残基表征这两路,不再反复迭代MSA。
Pairformer不是Evoformer换个名字那么简单。 AF2 Evoformer里把单链表征反向打回配对表征的外积均值模块没了。 沿MSA列方向的列注意力也没了。 AF3主干彻底不再维护MSA表征。 保留下来的是三角注意力、三角乘法这套对配对表征做几何约束的机制。 这个改动直接砍掉大量算力,也让模型对MSA质量差的输入更宽容,比如孤儿蛋白、人工设计蛋白。
第二个改动更彻底:IPA加FAPE整块换成扩散去噪。 这个逻辑得拆开说。
熟悉图像生成的人应该知道去噪扩散概率模型(DDPM)。 Stable Diffusion、DALL·E、Midjourney都用这套逻辑。 简单说就是:先给清晰照片一层层加高斯噪声,直到变成纯雪花。 再训练网络学怎么把雪花一点点擦掉,每擦一步恢复一点细节。 推理时给网络一张纯雪花,让它一步步擦,最后出来一张全新的照片。
AF3把这套逻辑搬到了原子坐标预测上。
训练时,给真实蛋白复合物的所有原子坐标加不同强度的高斯噪声。 低噪声让网络学局部几何,比如键长键角。 高噪声让网络学全局拓扑,比如结构域之间的位置关系。 网络的任务是,给定加噪后的坐标和Pairformer输出的配对表征,预测每个原子该往哪个方向回到真实位置。
推理时,先给一团纯噪声原子云,每个点带碳、氮、氧、磷等类型标签。 让网络一步步把这团云去噪成真实结构。
这个改动看起来只是换了个技术路径,但实现了三件AF2做不到的事:
第一是统一表征。 AF2的IPA专为氨基酸骨架定制。 AF3的扩散模块不管输入是氨基酸、核苷酸还是ATP里的一个碳,都当成带类型标签的原子云处理。
AF3内部把蛋白、核酸、小分子统一为两级表征:上层是token级,一个氨基酸或核苷酸算一个token,小分子和修饰部分每个原子单独算一个token。 下层是原子级,每个原子单独建模。 这套架构第一次让一个模型既能算蛋白-蛋白、蛋白-DNA、蛋白-小分子,还能算金属离子和糖基化修饰。
第二是几何不变性靠数据学习。 AF2的IPA是硬约束,网络结构本身保证SE(3)等变性。 AF3的扩散模块是软约束,网络不再显式等变。 靠训练时大量随机旋转、平移的数据增强,让网络自己学到等变特性。 代价是同一个输入旋转后,AF3可能给出略有不同的输出。 好处是网络更灵活,能容纳更多类型的分子。
第三是生成式特性带来不确定性,要多次采样再排序。 AF2一个输入对应一个确定输出。 AF3每次推理结果都不同,因为扩散过程本身有噪声。 默认做法是跑5个随机种子,每个种子采样5次,一共得到25个候选结构。 再用独立的置信度头给每个候选打分,指标包括pLDDT每残基置信度、PAE残基间误差预测、ipTM/PTM整体复合物对接质量。 最后按排名分数排序选最优。
这个排名分数不是黑箱。 AF3的GitHub文档公开了公式:0.8 × ipTM + 0.2 × pTM + 0.5 × disorder − 100 × has_clash。 界面质量ipTM权重0.8,整体结构pTM权重0.2。 只要任意两条链原子穿模超过100个,或者超过50%的链原子相撞,就触发穿模惩罚,直接扣100分。 分数范围在-100到1.5之间。
药企的BD、CADD团队要拿AF3输出做决策,得看ipTM是否超过0.8,也就是高质量界面。 pTM是否超过0.5,也就是整体折叠可信。 界面分低于0.6的基本要谨慎。 这套做法从图像生成领域搬来,采样多个候选再选最优,和AF2一次输出的思路完全不同。
训练还有几个细节。 AF3的训练数据截止到2021年9月30日。 PDB在那之后存的近3年新结构,都没进训练集。 训练用384个token的裁剪窗口,再微调到640、768个token,单卡 batch 256。 整套训练用TPU跑了数周,DeepMind没公开具体卡时。 推理门槛低很多,一块NVIDIA A100 80G就能跑4352个token以内的复合物。 学术圈拿到代码立刻就能本地复现。
AF3真的无所不能?
AF3上线那一周,整个药化圈都去官网跑自己的项目复合物。 把已知小分子配体放进去对接,看能不能复现已知的结合姿态。
跑出来的结果符合预期。 常规蛋白-蛋白对接比AlphaFold-Multimer 2.3好很多。 抗体-抗原界面平均DockQ从0.325升到0.422,提升约30%。 蛋白-DNA、蛋白-RNA预测效果比专门的核酸预测器好。 蛋白-小分子对接在PoseBusters评测里成功率约76%,比AutoDock Vina、Glide这些传统物理对接工具好。
这个数字一公布,计算机辅助药物设计(CADD)圈先躁动。 传统对接软件一年给药企算几百万次复合物,AF3的对接成绩如果稳定,整套商业模式都要重排。
但AF3不是没有能力边界。
第一个边界是生成幻觉。 无序区域、天然无序蛋白这类没有固定构象的序列,AF3经常会"编"出一个看似有序的螺旋或片层结构,还给出较高的pLDDT分数。 但这类序列在真实生物体内是软的,没有固定构象。 2024到2025年有好几篇论文专门量化这个问题。 也就是说,pLDDT高不等于结构正确,只等于模型自己认为正确。
第二个边界是评测基准的争议。 牛津大学Buttenschoen团队2024年在《化学科学》发了论文。 用308个2021年之后发布的蛋白-配体复合物做基准,除了RMSD≤2埃的要求,还加入物理合理性检查,重新评测AF3的小分子对接能力。
结论是:目标蛋白和PDBbind 2020训练集的最大序列相似度低于30%时,深度学习对接几乎拿不出同时满足2埃精度和物理合理性的姿态。 AF3的一部分对接能力,是识别训练时见过的相似配体。 这套评测还测出AF3有4.4%的立体化学违规率。
第三个边界是训练数据截止到2021年9月30日。 2021年10月之后实验解出的新折叠,比如特殊折叠的设计蛋白、新发现的天然蛋白家族,AF3都没见过。 要预测真正未知的新蛋白,AF3也没把握。
第四个边界是不输出亲和力,也不模拟动力学。 AF3只给结构坐标和置信分,不给结合常数、自由能、结合解离速率,也不输出分子动力学轨迹。 相分离、构象切换、变构调控、磷酸化导致的功能态切换这些问题,AF3都回答不了。 2025年6月MIT团队发布Boltz-2,专门把结合亲和力当成显式任务做,就是补AF3的缺口。
监管端截至2026年5月,公开渠道没有明确表态,认可AF3计算结构作为IND申报的独立支持证据。 当前AF3在监管层面更适合做假设生成和结构辅助,不能直接当申报材料。
第五个边界是商业访问限制。 AlphaFold Server免费但限速,每个学术账号每天最多跑30个任务,单任务上限5000个token。 不预测水分子、氢原子,也不感知膜平面。 膜蛋白、金属配位水、氢键网络这些结果,不要指望官网服务器直接输出。
服务条款明确禁止商业用途,禁止用输出训练其他结构预测模型,禁止把输出导入对接、虚拟筛选工具。 代码2024年11月上传GitHub,许可证是CC BY-NC-SA 4.0。 权重需要学术身份单独申请,不能二次分发。 商业访问权由同构实验室独家持有。

30亿美刀押注新赛道
商业化端的节奏非常快。 2024年1月7日,同构实验室一天签了两笔合作。 礼来预付4500万美元,里程碑上限17亿美元。 诺华预付3750万美元,里程碑上限12亿美元。 两笔合计潜在总金额约30亿美元,这是上限不是已到账收入。
2026年5月12日,Thrive Capital领投21亿美元B轮融资。 Alphabet和GV跟投,MGX、淡马锡、CapitalG、英国主权AI基金新进。 这笔钱将用于升级IsoDDE药物设计引擎、全球扩张、推进自有管线进入临床。 公司公开表示,2026年内要把首个AI设计的小分子推进I期临床。
AF3的出现,对国内AI制药公司来说五味杂陈。
一方面,一个模型搞定全类生物分子结构的路径已经被验证。 各家AI制药公司原来靠AF2加自研对接模块拼出来的管线价值缩水。 你能做的,AlphaFold Server开个标签页也能做。
另一方面,因为商业限制,国产开源复现成了必须走通的路。 2024年8月,百度螺旋桨PaddleHelix团队推出HelixFold 3,号称全球首个完整复现AF3的开源版本。 在小分子、核酸、蛋白互作上的指标对标DeepMind。
2026年2月,字节跳动旗下的Protenix-v1以Apache 2.0协议全开源,代码和权重都开放。 在和AF3同等训练数据截止时间、同等模型尺寸的前提下,指标超过AF3。 两个月后又推出Protenix-v2,参数464M,抗体-抗原对接和小分子合理性指标再提升一档。 这两条国产路线,已经把AF3权重不公开的护城河填平了,至少在学术和开源使用层面是这样。
海外开源端也动作不断。 MIT Jameel Clinic 2024年11月发布Boltz-1,MIT协议全开源。 Chai Discovery 2024年9月发布Chai-1,非商用研究开源。 Meta FAIR的ESMFold走另一条技术路线,不需要MSA,靠蛋白语言模型预测。 2025年各家又推出下一代版本:Boltz-2补上了亲和力预测能力,Chai-2把零样本抗体设计的命中率推到16%。 2024到2025年这一年半,开源生态几乎补齐了AF3的所有能力。
磨结构的苦少了大半
我师兄花四年做出来的膜蛋白结构,现在用AlphaFold Server跑一次只要5分钟。 但他做的那些事,长晶体、解相位、跑动力学、看构象变化,AF3还是回答不了。 AF3给出的结果,是2021年9月之前的训练分布里,这个序列最可能的折叠形态。 这件事非常有用,而且几乎免费。 但AF3不会告诉你,这个蛋白在细胞膜里、有配体存在时、37度生理盐水中、磷酸化之后是什么形态。天然无序蛋白、构象动力学、膜蛋白真实环境、全新折叠,这几个问题还没解决。
安芬森1972年提出的序列决定结构是对的。 莱文索尔1969年提出的搜索空间太大也是对的。 AF3没有绕过莱文索尔悖论。 它把PDB60年沉淀的二十多万个实验结构里的先验,压缩进了几亿参数。 神经网络预测新序列时,本质是在找以前见过的最相似的结构。
下次再看到博士生为了一个结构泡三年实验室,可以提醒他一句: 先去AlphaFold官网跑一遍,至少能省下养细胞的两年时间。
