 夜雨聆风
夜雨聆风今天我们学习软件SibeliaZ的原理安装以及使用方法。
在GitHub上,最新版本为 SibeliaZ 1.2.7 ,最后更新于25年7月7日。作者是宾夕法尼亚州立大学的 Ilia Minkin 和 Paul Medvedev。
https://github.com/medvedevgroup/SibeliaZ
SibeliaZ的简介
SibeliaZ 是一款全基因组比对与共线性局部区块构建流程工具。其区块坐标以 GFF 格式输出,比对结果则采用 MAF 格式。该工具适用于多个相似度较高的基因组数据,例如同一物种的不同菌株。当数据集满足从序列末端到最近共同祖先的位点替换率不超过 0.09(即 9 个 PAM 单位)时,SibeliaZ 运行效果最佳。目前该工具暂不支持长度超过 4294967296 碱基对(bp)的染色体序列(官方指出该问题将在后续版本中修复),不过这个长度对于处理细菌的基因组已经足够的了。
SibeliaZ基于de Bruijn图算法,通过识别基因组间的保守k-mer锚定同源区域,逐步拼接扩展为局部共线性区块,通俗来说,就是先在所有基因组中找共同的短序列作为‘定位锚点’,再将相邻锚点拼接扩展为完整的同源共线性区块,最终完成多基因组全序列比对,适配近缘物种及菌株的高精度比对需求。
SibeliaZ的安装
SibeliaZ的官方给出两种安装方法。
安装SibeliaZ最简单的方法是使用bioconda。 安装好Bioconda环境后,安装 sibeliaz 包:
conda install sibeliaz如果要自行编译代码,需要最近安装以下代码 软件(仅限Linux):
Git
CMake
一个支持 C++11 的 GCC 编译器
ZLIB库(针对SPOA对齐器)
后续再进行 SibeliaZ 源码下载解压编码配置环境变量等工作。这里我们推荐使用 conda 进行安装,因为这不仅更简单,而且不会有版本冲突环境崩溃等问题,特别是对新手,如果您没有学会使用conda,非常推荐。
SibeliaZ的使用
SibeliaZ 接收一组 FASTA 文件作为输入。最简单的运行 SibeliaZ,为以下命令:
sibeliaz <input FASTA files>例如:
sibeliaz genome1.fa genome2.fa 比对结果会依据序列ID生成,因此FASTA文件标题行中的所有输入序列 必须拥有唯一ID。对于这一个 唯一ID ,需要准确理解,这不仅指一个fa文件里的多条序列ID不能有重复,对于你输入到 SibeliaZ 的所有fa文件(无论是两个,还是一百一千个),只要是 > 后面的ID,都不能重复。这一点真的很重要,否则你的分析是会进行报错的。
默认输出至当前工作目录下的 sibeliaz_out 文件夹,内含记录区块坐标的 blocks_coords.gff文件,以及存储比对结果的 alignment.maf 文件。
SibeliaZ配有多项参数,可调节精度与运行效率,下文将逐一说明。
SibeliaZ 的输出
输出目录包含以下文件:
blocks_coords.gff:记录局部共线性区块坐标。ID 字段相同的行,代表同一区块的不同拷贝。
alignment.maf:输入基因组的全基因组比对结果。
注意:最终比对是对局部共线性区块进行全局比对,该过程内存占用极高。部分区块(尤其拷贝数量多、序列较长时)可能因内存不足比对失败,大内存设备也可能出现此问题。 输出目录下的 blocks 子文件夹,存放所有比对失败区块的 FASTA 文件,每个文件对应一个区块及其全部拷贝。FASTA 标题行记录各拷贝坐标,格式同 MAF 条目,字段以分号分隔。
若后续分析无需比对结果,可使用 -n 参数跳过比对步骤,仅输出区块坐标。此时程序不会生成。
构建共线性块
你还可构建部分分析所需的长共线性区块。SibeliaZ 默认会附带安装由Mikhail Kolmogorov编写的 maf2synteny 工具。若已有 SibeliaZ 输出结果,可直接用该工具处理。
maf2synteny <GFF or MAF file created by SibeliaZ>一般来说,这是一整套构建共线性块的流程,可以通过调整 maf2synteny 参数大小,构建符合自己数据的大小不同的共线性块。比较只用 SibeliaZ 构建的块,还是太碎太小。
影响 SibeliaZ 的准确性的参数
k 值
该参数设定德布鲁因图的阶数,用于权衡比对灵敏度、运行速度与内存占用。
-k <an odd integer>通常 k 值越小,比对灵敏度越高,但运行速度越慢。细菌等小型基因组建议设为 15,哺乳动物级别的大型基因组建议设为 25,默认值为 25。
气泡长度阈值
SibeliaZ通过检索图中的长气泡链开展分析。气泡指起止点相同的两条路径,长气泡链一般对应同源序列。但若气泡内路径过长,易产生假同源匹配。
工具会丢弃路径长度超出阈值的气泡,对应参数:
-b <integer>默认值为 200。增大该值可识别更多分化程度较高的序列,但取值过高会降低比对准确度。
局部共线块大小
SibelaZ 只输出超过指定阈值的块,该阈值由
-m <integer>默认值是50。警告:提高该参数可能会显著增加 减缓计算速度。
SibeliaZ的技术参数
跳过比对
如需仅输出区块坐标、跳过比对流程,使用参数:
-n线程数
设置程序可调用的最大线程数,参数格式:
-t <integer>程序默认会占用全部可用线程,你可通过该参数限制线程数量。流程不同阶段的并行能力存在差异:TwoPaCo 最多仅使用16个线程,而图谱分析模块与全局比对模块会充分调用所有线程。
内存分配
图谱构建工具 TwoPaCo 会为布隆过滤器预分配内存。默认情况下,布隆过滤器容量为输入文件总大小的三倍。可通过以下参数手动指定容量:
-f <memoryamountinGB>输出目录
自定义结果文件的存放目录,参数格式:
-o <directory>默认输出至当前工作目录下的 sibeliaz_out 文件夹。
实例
先是利用conda安装 SibeliaZ,验证安装成功
# 创建名为 sibeliaz_env 的环境,指定 python3.8(通用兼容版本)conda create -n sibeliaz_env python=3.8 -y
#激活环境conda activate sibeliaz_env#激活成功后,终端前缀会出现 (sibeliaz_env)
#配置 Bioconda 源(首次使用必须配置)conda config --add channels biocondaconda config --add channels conda-forgeconda config --set channel_priority strict
# 安装软件conda install sibeliaz -y# 验证是否安装成功sibeliza -h# 如果输出为 Usage: [-k <odd integer>] [-b <integer>] [-m <integer>] [-a <integer>] [-t <integer>] [-f <integer>] [-o <output_directory>] [-n] <input file># 则表明安装成功
使用SibeliaZ分析数据
sibeliaz -k 15 -n -o salmonella_synteny_out fixed_assemblies/*.fasta确定好 SibeliaZ 安装成功后,使用这行命令即可使用 SibeliaZ 分析数据。
其中我的数据是细菌的染色体测序结果,所以使用 -k 15,使用 -n 跳过序列比对,使用 -o 指定输出目录,同时后面使用一个正则匹配把所有的fa文件一起输入分析,供大家参考。各参数意义及使用方法前面已经讲解。
SibeliaZ 输出结果解读
输出结果在文件夹 salmonella_synteny_out 里,名为 blocks_coords.gff
内容如图:
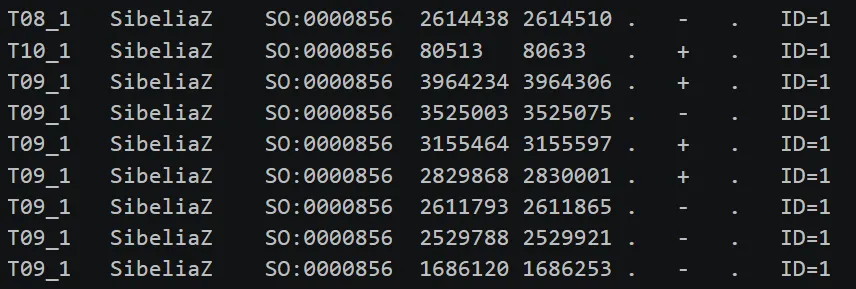
文件解读,如下表:
T08_1T10_1 / T09_1,代表不同的基因组 | ||
SibeliaZ | ||
SO:0000856sequence_feature,表示序列特征 | ||
. | ||
+- 负链,代表该同源片段的方向 | ||
. | ||
ID=1 |
maf2synteny构建共线性块
运行下面代码,构建不同大小的共线性块。
maf2synteny -b 500 salmonella_synteny_out/blocks_coords.gff-b 后面跟不同阈值构建不同大小的块,输出结果如下图:
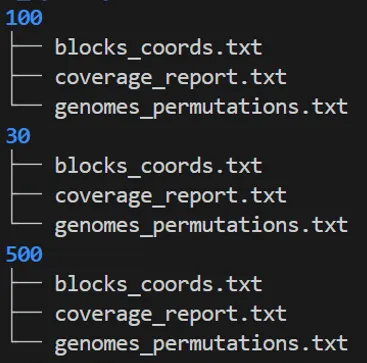
每个阈值文件夹(30/、100/、500/)里的文件,对应 maf2synteny 处理后的结果,解读如下表:
blocks_coords.txt | |
coverage_report.txt | |
genomes_permutations.txt |
blocks_coords.gff 是 SibeliaZ 直接输出的原始数据,而 blocks_coords.txt 是 maf2synteny 整理后的、更适合下游工具使用的格式到这里,我们了解了 SibeliaZ 的一些背景和原理,学习了SibeliaZ 的安装使用参数调整以及后续处理流程,并且我们还提供了一个实例,想真的学会理解透一个软件,还是得自己动手去找点数据处理一下,实践出真知,如果您在使用软件时遇到问题,欢迎在评论区留言或者私信我们,一起交流学习!
后期我们会陆续推出更多生信软件的教程,期待您的关注。如果您在文献复现以及生信分析时遇到问题,也欢迎联系我们一起交流学习!
多看多学多做,如果您觉得我们写的推文还可以,请给我们点个赞,这是我们前进的最大动力,真心感谢!
